
Abstract
Chromobacterium sp. strain DS-1 produces an extracellular cholesterol oxidase that is very stable at high temperatures and in the presence of organic solvents and detergents. In this study, we cloned and sequenced the structural gene encoding the cholesterol oxidase. The primary translation product was predicted to be 584 amino acid residues. The mature product is composed of 540 amino acid residues. The amino acid sequence of the product showed significant similarity (53–62%) to the cholesterol oxidases from Burkholderia spp. and Pseudomonas aeruginosa. The DNA fragment corresponding to the mature enzyme was subcloned in the pET-21d(+) expression vector and expressed as an active product in Escherichia coli. The cholesterol oxidase produced from the recombinant E. coli was purified to homogeneity. The physicochemical properties were similar to those of native enzyme purified from strain DS-1. K m and V max values of the cholesterol oxidase were estimated from Lineweaver–Burk plots. The V max/K m ratio of the enzyme was higher than those of commercially available cholesterol oxidases. The circular dichroism spectral analysis of the recombinant DS-1 enzyme and Burkholderia cepacia ST-200 cholesterol oxidase showed that the conformational stability of the DS-1 enzyme was higher than that of B. cepacia ST-200 enzyme at higher temperatures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
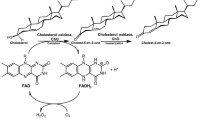
Cholesterol oxidase (EC 1.1.3.6) is a flavin adenin dinucleotide (FAD)-dependent enzyme that in most cases catalyzes the oxidation of cholesterol (cholest-5-en-3β-ol) using oxygen as an electron acceptor to form cholest-4-en-3-one (CEO) and hydrogen peroxide (Smith and Brooks 1974; Uwajima et al. 1974). On the other hand, some cholesterol oxidases from Burkholderia cepacia strain ST-200, Pseudomonas spp., and Chromobacterium sp. strain DS-1 oxidize cholesterol to 6β-hydroperoxycholest-4-en-3-one (HCEO) but not the CEO produced by most cholesterol oxidases (Doukyu and Aono 1999; Doukyu et al. 2008). Cholesterol oxidases have been isolated from various microorganisms such as Arthrobacter (Liu et al. 1988), Brevibacterium (Uwajima et al. 1974), Burkholderia (Doukyu and Aono 1998), Chromobacterium (Doukyu et al. 2008), Cellulomonas (Srisawasdi et al. 2008), Corynebacterium (Shirokane et al. 1977), Mycobacterium (Smith et al. 1993), Nocardia (Richmond 1973), Pseudomonas (Lee et al. 1989), Rhodococcus (Johnson and Somkuti 1991), Schizophyllum (Fukuyama and Miyake 1979), Streptomyces (Tomioka et al. 1976), and Streptoverticillium (Inouye et al. 1982).
With only the exception of glucose oxidase, cholesterol oxidase is the most widely used enzyme in clinical laboratories. It is used for the determination of cholesterol concentrations in serum and other clinical samples and as a probe to study membrane structure (MacLachlan et al. 2000). In addition, cholesterol oxidase shows insecticidal activity that is a vital part of pest control strategies employing transgenic crops (Cho et al. 1995; Purcell et al. 1993). Moreover, cholesterol oxidase has been used for the optical resolution of non-steroidal compounds and allylic alcohols (Biellmann 2001; Dieth et al. 1995) and for the bioconversion of a number of 3β-hydroxysteroids in the presence of organic solvents (Aono and Doukyu 1996; Aono et al. 1994; Kazandjian et al. 1986). Since cholesterol is an insoluble compound, detergents or organic solvents are often added to the reaction solution to act as a solubilizer, and because detergents or organic solvents often inactivate cholesterol oxidases as well as most enzymes (Doukyu and Aono 2001; Isobe et al. 2003; Pollegioni et al. 1999), a detergent- and organic solvent-tolerant cholesterol oxidase would be useful for the applications described above. A detergent- and organic solvent-tolerant cholesterol oxidase has been reported from B. cepacia strain ST-200 (Doukyu and Aono 1998; Doukyu and Aono 2001), and a detergent-tolerant cholesterol oxidase has been reported from γ-Proteobacterium Y-134 (Isobe et al. 2003).
We previously reported the screening and purification of an extracellular cholesterol oxidase from Chromobacterium sp. strain DS-1 and listed some of its properties (Doukyu et al. 2008). This was the first report of cholesterol oxidase from the genus Chromobacterium. Most cholesterol oxidases from various bacterial sources such as Brevibacterium sp., Streptomyces sp., Cellulomonas sp., Nocardia sp., Nocardia erythropolis, Pseudomonas fluorescens, and B. cepacia ST-200 lost most of their activity after incubation for 30 min at 60–80°C (Doukyu and Aono 2001; Doukyu et al. 2008). By contrast, the enzyme from strain DS-1 retained 80% of its original activity even at 85°C after 30 min. As far as we can tell, cholesterol oxidase from strain DS-1 exhibits the highest thermal stability among all cholesterol oxidases reported so far. Furthermore, DS-1 cholesterol oxidase was more stable in the presence of various organic solvents and detergents than commercially available cholesterol oxidases, including a detergent- and organic solvent-tolerant cholesterol oxidase from B. cepacia ST-200. Cholesterol oxidase from strain DS-1 oxidized cholesterol to HCEO. A gene sequence of an HCEO-forming cholesterol oxidase has been reported only from B. cepacia strain ST-200 (Doukyu and Aono 2001); no other gene sequence of HCEO-forming cholesterol oxidase has been reported so far.
In this paper, we report the cloning of a cholesterol oxidase gene from Chromobacterium sp. strain DS-1, expression of the gene in Escherichia coli, and the purification and partial characterization of the enzyme produced by the recombinant E. coli. This is the first report concerning cloning of a gene encoding cholesterol oxidase from the genus Chromobacterium.
Materials and methods
Chemicals and enzymes
ExTaq polymerase and PrimeSTAR HS DNA polymerase were purchased from Takara Bio (Kyoto, Japan). Commercially available cholesterol oxidases were obtained as follows: N. erythropolis (Fluka, Steinheim, Germany), Nocardia sp. (Calbiochem, San Diego, CA, USA), P. fluorescens (Sigma Chemicals, St Louis, MO, USA), Streptomyces sp. SA-COO (Toyobo, Tsuruga, Japan). Cholesterol oxidase from B. cepacia strain ST-200 was purified as described previously (Doukyu and Aono 2001) and used in this study. Triton X-100 was purchased from Sigma Chemicals; Tween 20, sodium dodecyl sarcosinate (sarcosyl), and sodium dodecyl sarcosinate (SDS) were purchased from Wako Pure Chemical Industries, Osaka, Japan; and sodium polyoxyethylene alkyl (C12–13) ether sulfate (Emal 20CM) was obtained from Kao Corp., Tokyo, Japan. Organic solvents used were of the highest quality commercially available.
Strains and media
Chromobacterium sp. strain DS-1 (Doukyu et al. 2008) was used as the source of DNA encoding the cholesterol oxidase. The vector plasmid, pHSG398, was purchased from Takara Bio (Kyoto, Japan). pBluescript II SK+ (pBSII) was purchased from Toyobo Biochemical (Osaka, Japan). pET-21d(+) was produced by Novagen (Madison, WI, USA). E. coli DH5α (supE44, ΔlacU169(ϕ80lacZΔM15), hsdR17, recA1, endA1, gyrA96, thi-1, relA1) was used for cloning and expression of the gene. E. coli Rosetta (DE3) pLysS [F−, ompT, hsdSB(RB − mB −), gal, dcm, lacYI (DE3), pLysSRARE (CamR)] (Novagen) was used for expression of the gene. E. coli strains were grown in modified Luria broth (LB medium) consisting of 1% Bacto tryptone (Difco Laboratories, Detroit, MI, USA), 0.5% Bacto yeast extract (Difco), and l% NaCl. When necessary, the medium was solidified with 1.5% (wt vol−1) agar and supplemented with appropriate antibiotics. LB agar medium supplemented with 64 mM sodium cholate, 0.3% Triton X-100, and 0.9 mM cholesterol (LBC agar medium) was also used for the isolation of a transformant containing the cholesterol oxidase gene.
Amino acid sequence
Extracellular cholesterol oxidase from strain DS-1 was purified from the culture supernatant (Doukyu et al. 2008).The amino acid sequence was determined using an automated protein sequencer (model G1005A; Hewlett-Packard, Palo Alto, CA, USA). For determination of the amino acid sequence of internal regions of the cholesterol oxidase, the purified enzyme was digested with lysyl endopeptidase from Achromobacter lyticus M497-1 (Wako Pure Chemical Industries). The digestion mixture was analyzed by SDS-PAGE. Peptides were electroblotted from the gel onto a polyvinylidene difluoride membrane (Immobilon; Millipore, Bedford, MA, USA). The N-terminal amino acid sequences of selected peptides were determined as described above.
Genetic analysis
DNA manipulations, including preparation of plasmids, restriction enzyme digestion and ligation, and transformation of E. coli, were carried out by standard methods (Sambrook et al. 1989). Southern hybridization was performed by means of a DNA labeling and detection kit (Roche, Basel, Switzerland). Nucleotide sequences of the cloned DNA fragments were determined with a DNA sequencing system (Prism 377; Perkin-Elmer Applied Biosystems, Foster City, CA, USA). The dideoxy chain termination method was used to sequence the gene. The DNA and the predicted protein sequences were analyzed using the BLAST (Altschul et al. 1990) network service from the National Center for Biotechnology Information. The phylogenetic tree based on the comparison of deduced amino acid sequences was constructed using the neighbor-joining method with GENETYX software (GENETYX Co., Tokyo, Japan).
PCR conditions for cloning of the cholesterol oxidase gene
PCR primers, primer 1, 5′-CCIAA(C/T)AA(C/T)TT(C/T)CCIGCIGA(A/G)AT(A/C /T)CC-3′ and primer 2, 5′-GTIACIC(T/G)IA(A/G)IGTIGTIGG(C/T)TTIAC(A/G)TA-3′, were designed based on N-terminal and internal amino acid sequences of the cholesterol oxidase, respectively. A DNA fragment (1 kb) was amplified from the chromosomal DNA of strain DS-1 by PCR using ExTaq polymerase and the primers. Amplification (30 cycles of 1 min at 96°C for denaturation, 1 min at 45°C for annealing, 1 min at 72°C for extension) was carried out using the GeneAmp PCR system 2400 (Perkin-Elmer Applied Biosystems). The amplified fragment was blunted and ligated into the SmaI site of pHSG398.
For identification of the entire nucleotide sequence of the cholesterol oxidase gene, the chromosomal DNA was digested with SmaI and ligated with T4 DNA ligase to form circularized DNA. Then, inverse PCR was performed to amplify a fragment using ExTaq polymerase, the SmaI-self-circularized DNA molecule as a template, and appropriate primers P-S, 5′-GACAACCTGCCCGACGAAGT-3′ and P-AS, 5′-CGCACCTTATAACCGTTGTC-3′. These primers were designed based on the nucleotide sequence of the DNA insert ligated into the SmaI site of pHSG398. The PCR conditions were as follows: 1 min at 94°C for denaturation, 1 min at 50°C for annealing, 2 min at 72°C for extension, for 30 cycles.
For isolation of the entire cholesterol oxidase gene, primers were designed according to the determined sequence. PCR was performed to amplify the fragment, including the entire cholesterol oxidase gene using PrimeSTAR HS DNA polymerase with high fidelity, genomic DNA as the template, and primers CO-S, 5′-GGTCTAGATAGCTAAATCAGCGATCCGC-3′ (XbaI site underlined) and CO-AS, 5′-GGGGTACCCGGCGAATACCGCTTCGAAA-3′ (KpnI site underlined). The PCR conditions were as follows: 1 min at 94°C for denaturation, 1 min at 52°C for annealing, 2 min at 72°C for extension, for 30 cycles. The amplified fragment was inserted into the same sites in pBSII SK+.
Construction of pETcox
The cholesterol oxidase gene was amplified from pBScox by designing a set of primers with incorporated restriction enzyme sites NcoI/BamHI (Fig. 1). The primers COR-S: 5′- TACCATGGCTACTTGCAGCCAACCCAATAA -3′ (NcoI site underlined) and COR-AS: 5′-AAGGATCCTTAGCTCGCGAGATCGTAAA -3′ (BamHI site underlined) were used to clone a DNA fragment encoding the complete cholesterol oxidase gene except the signal peptide. As a consequence of the introduction of NcoI site, the recombinant enzyme contained an extra Met-Ala at the N-terminal end. The amplified gene and plasmid pET-21d(+) were digested with the same restriction enzymes, NcoI and BamHI. The ligated plasmid (pETcox) was used to transform the E. coli Rosetta strain.

Scheme of the construction of pETcox derived from the expression vector pET21-d(+)
Purification of the recombinant cholesterol oxidase
The E. coli Rosetta strain harboring pETcox was grown in 100 ml of LB medium supplemented with 100 μg ml−1 ampicillin and 25 μg ml−1 chloramphenicol on a rotary shaker (120 rpm) at 30°C. The expression of the cholesterol oxidase gene was induced by adding 1 mM of isopropyl-β-d-thiogalactopyranoside (IPTG) when the cell density reached an A 600 of ca. 0.5. After incubation during 12 h at 30°C, the cells were harvested by centrifugation (6,000×g, 15 min, 4°C) and resuspended with 10 ml of 10 mM Tris–HCl (pH 8.0). The resuspended cells were lysed using an ultrasonic disruptor UD-200 (Tomy Seiko, Tokyo, Japan) at 60 W intermittently in an ice bath. The sonicated lysate was cleared by centrifugation (10,000×g, 10 min, 4°C), and then the cleared cell lysate was heated at 70°C for 30 min and centrifuged (8,000×g, 20 min, 4°C). The supernatant was loaded on a column (2.5 by 8 cm) of DEAE-cellulose DE52 (Whatman, Maidstone, England) equilibrated with the Tris–HCl buffer. The column was washed with 100 ml of the Tris buffer and then eluted with a linear gradient of NaCl concentrations of 0 to 250 mM in 400 ml of the Tris buffer. The fractions with the cholesterol oxidase activity were pooled.
Determination of protein concentration
The protein concentration was determined using the method of Bradford (1976) and bovine serum albumin as the standard.
SDS-polyacrylamide gel electrophoresis of proteins
Samples were dissolved in a solution containing 1% (wt vol−1) sodium dodecyl sulfate (SDS), 2.5% (vol vol−1) β-mercaptoethanol, 30% (vol vol−1) glycerol, and 30 mM Tris–HCl (pH 6.8) and heated in a boiling water bath for 5 min. The samples were run on a SDS-polyacrylamide gel as described by Laemmli (1970).
Assay of the cholesterol oxidase activity
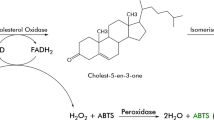
Cholesterol oxidation activity was assayed by measuring H2O2 generation accompanying the oxidation of cholesterol as described previously (Doukyu and Aono 1998). One unit of enzymatic activity was defined as the amount required to oxidize 1 μmol cholesterol min−1 at 30°C. Unless otherwise stated, this is the method we used for the determination of enzyme activity in this study. To investigate the optimum pH for the enzyme activity, we estimated the cholesterol oxidase activity by monitoring the consumption of oxygen, and to investigate the optimum temperature for the enzyme activity, we estimated the cholesterol oxidase activity by measuring the A 249 of λ max value of HCEO, as described previously (Doukyu et al. 2008).
Determination of K m and V max values of the cholesterol oxidase
Cholesterol oxidation activity was assayed by measuring H2O2 generation. The K m and V max values were estimated from Lineweaver–Burk plots of data obtained with the assay solution containing 0 to 1 mM cholesterol.
CD spectral analysis of the cholesterol oxidase
The circular dichroism (CD) spectra analysis of the cholesterol oxidase was conducted using a J-820 spectropolarimeter (JASCO, Tokyo, Japan). The measurement of CD spectra from 195 to 250 nm was performed at each temperature (60–95°C at 5°C intervals) using a 0.1-cm path-length cell. The concentration was set at 0.2 mg ml−1. The top of the cell was completely closed using a cap to minimize evaporation. Data pitch, bandwidth, response, scanning speed, and accumulation were set at 0.1 nm, 1 nm, 8 s, 50 nm per min, and four times, respectively. The secondary structure was analyzed using three component model (α-helix, turn, and random coil) reference spectra (Yang et al. 1986).
Nucleotide sequence accession number
The nucleotide sequence of the cholesterol oxidase gene of strain DS-1 has been deposited in the GenBank/EMBL/DDBJ database under the accession number AB456533.
Results
Cloning of the cholesterol oxidase gene
The N-terminal amino acid sequence of the cholesterol oxidase purified from culture supernatant was TCSQPNNFPAEIPLYKQSFKN as described previously (Doukyu et al. 2008). The enzyme was cleaved with lysyl endopeptidase, and the resulting peptides were fractionated by SDS-PAGE. Among several peptides separated on the gel, the N-terminal amino acid sequence of 22 kDa was determined. The sequence obtained was NLLLYVKPTTLRVTANGYAV. The sense primer (primer 1) and the antisense primer (primer 2) were designed based on the determined N-terminal amino acid sequences of the mature enzyme and the 22-kDa peptide, respectively. Inosine was incorporated into the primers at positions of four-base redundancy to reduce the complexity of the primer mixtures. A DNA fragment of about 1 kb was amplified from the chromosomal DNA of strain DS-1 by PCR with a combination of primer 1 and primer 2. The amplified fragment was ligated into the SmaI site of pHSG398. This plasmid was designated pHSGcox. The nucleotide sequence of the insert was analyzed to confirm that the amplified DNA corresponded to the determined amino acid sequences.
The DNA inserted in pHSGcox was digested with BamHI and SacI at both of the sites derived from the original plasmid pHSG398. The excised fragment (1 kb) was recovered and labeled with digoxigenin. Chromosomal DNA of DS-1 was digested with various restriction enzymes and analyzed by Southern hybridization, using the digoxigenin-labeled DNA as a probe, to assist in the choice of restriction enzymes for the inverse PCR. The results allowed us to create a physical map of the vicinity of the cholesterol oxidase gene (data not shown).
Chromosomal DNA of strain DS-1 digested with SmaI showed a single hybridization band of 3 kb. DNA fragments of 2–4-kb long were recovered from the SmaI digest of the chromosomal DNA and were ligated to form a self-circularized DNA molecule. A DNA fragment of 2.1 kb was amplified from the self-circulated molecules as the template and primers P-S and P-AS by inverse PCR. The sequence of the 2.1-kb DNA fragment included the sequence of 5′ and 3′ flanking regions of the cholesterol oxidase gene. The fragment (2.0 kb) containing the entire cholesterol oxidase gene was amplified using chromosomal DNA as the template and the primers CO-S and CO-AS and inserted into the XbaI/KpnI sites of pBSII in the same direction as Plac of the vector. E. coli DH5α was transformed with the resulting plasmids, pBScox. A clone, grown on LBC agar containing ampicillin, formed a turbid halo around its colony (Fig. 2) and contained a recombinant plasmid with a 2.0-kb XbaI/KpnI insert. The halo was formed because of the low solubility of HCEO generated by the cholesterol oxidase from cholesterol, as described previously (Doukyu and Aono 2001; Doukyu et al. 2008).

Turbid halo around colonies of E. coli DH5α carrying pBScox. E. coli DH5α cells carrying pBluescript II SK+ (left side) or pBScox (right side) were grown on LBC agar containing 50 μg ml−1 ampicillin at 30°C overnight
Nucleotide sequence of the cholesterol oxidase gene
The nucleotide sequence of the cholesterol oxidase gene contains an open reading frame of 2,004 bp within a TAG at nucleotide position 295 and a TAG codon at nucleotide position 2,164 (Fig. 3). We found a structural gene of 1,755 bp, coding for a polypeptide consisting of 584 amino acid residues, with the ATG initiation codon at nucleotide position 412 and the TGA termination codon at position 2,164. A potential ribosome-binding site (GGAG) was observed seven bases upstream of this ATG. The nucleotide sequence of the structural gene coded for the N-terminal and the internal amino acid sequences that were determined for the cholesterol oxidase purified from DS-1. The N-terminal amino acid sequence showed that the signal peptide was proteolytically processed between Leu-44 and Thr-45 upon excretion and secretion in strain DS-1. The mature polypeptide consisted of 540 amino acid residues, with a molecular mass of 58,424 Da. This is nearly 58 kDa, the mass of the native enzyme as previously estimated by SDS-PAGE (Doukyu et al. 2008).

Nucleotide sequence of the cholesterol oxidase gene from strain DS-1 and the deduced amino acid sequence. Numbers on the right denote nucleotide positions and amino acid positions. The N-terminal and the internal amino acid sequences determined through analysis of the mature cholesterol oxidase from strain DS-1 are underlined with dotted lines. The vertical arrowheads indicate the cleavage sites of the enzyme in strain DS-1. The Shine–Dalgarno sequence (SD) is underlined. A putative histidine residue (His107) that was covalently bound to a FAD cofactor is enclosed in a circle. A putative glutamic acid residue, Glu445, that may act as the base for both the oxidation and the isomerization steps of the catalytic reaction is enclosed in a shadowed box. Putative hydrophilic amino acid residues lining the cavity near the FAD cofactor (Glu299, Arg447, Glu518, and Lys524) are enclosed in a box, respectively
Sequence comparison of cholesterol oxidases
We found several amino acid sequences that showed significant similarities to DS-1 cholesterol oxidase via a BLAST search. An alignment of the amino acid sequence of DS-1 cholesterol oxidase with corresponding sequences from other bacterial cholesterol oxidases was used to construct the phylogenetic tree (Fig. 4). DS-1 cholesterol oxidase displayed relatively high similarity to the cholesterol oxidases (53–62%) from Burkholderia spp. and Pseudomonas aeruginosa strains (Table 1). A lower similarity was found in cholesterol oxidases from Streptomyces ambofaciens (47%), Rhodococcus erythropolis (44%), and Brevibacterium sterolicum (42%).

Phylogenetic tree of homologues of the cholesterol oxidase from strain DS-1. A phylogenetic tree based on the comparison of the deduced amino acid was constructed using the neighbor-joining method. The scale bar represents 0.1 substitutions per amino acid position. Bootstrap values greater than 40% are shown as percentages on branches
Expression and purification of the recombinant cholesterol oxidase
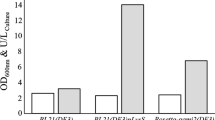
Overexpression of the recombinant cholesterol oxidase was attempted by using the pET system. The nucleotide sequence of DS-1 cholesterol oxidase contained codons (ten of Pro codon ccc, four of Ile codon gga, one of Arg codon agg, and one of Arg codon cga) rarely used in E. coli. The translation of a gene might be limited by the codon usage. Therefore, we used an E. coli Rosetta host strain, BL21 derivative, that was able to supply tRNAs for these rare codons on a compatible plasmid. We confirmed that there was no mutation in the cholesterol oxidase gene on the pETcox by resequencing. The cholesterol oxidase activity of the sonicated lysate of E. coli Rosetta harboring pETcox grown on LB medium supplemented with antibiotics and IPTG was 2.28 U mg−1. This activity was about fivefold higher than that (0.45 U mg−1) of E. coli DH5α harboring pBScox grown under the same conditions. Table 2 summarizes the purification steps used. High thermal stability of DS-1 enzyme made it possible to easily remove most proteins from E. coli by the heat treatment at 70°C. The cholesterol oxidase activity passed through the DE52 column. This step was effective for purifying the enzyme. In the end, the enzyme was purified 7.3-fold from the cell lysate. The specific activity of the purified cholesterol oxidase was 16.7 U mg−1 of protein. This value was slightly higher than that observed for the purified enzyme of strain DS-1 (13.9 U mg−1; Doukyu et al. 2008). The purified preparation gave a single band by SDS-PAGE (Fig. 5). The molecular mass was estimated to be 58 kDa. The N-terminal amino acid sequence of the expressed protein was ATCSQPNN. This result indicated that the methionine of the N-terminal of the recombinant enzyme seemed to be removed by methionyl aminopeptidase of E. coli.

SDS-PAGE of cholesterol oxidase during the purification procedure. Samples containing 0.04 U of cholesterol oxidase were applied on SDS-12.5% (wt vol−1) polyacrylamide gel. The gel was stained with Coomassie Brilliant Blue R-250 (CBB). Lane M molecular size markers (kilodaltons), lane 1 cleared sonicated lysate, lane 2 cleared lysate after the heat treatment at 70°C, lane 3 fraction from the DEAE-cellulose DE52 column
Prosthetic groups
The recombinant enzyme solution was yellow, like a typical flavoprotein, exhibiting two absorption maxima, at 355 and 450 nm. Most cholesterol oxidases contain 1 mol of FAD per mole of protein as a prosthetic group (Coulombe et al. 2001; Doukyu and Aono 1998; Kamei et al. 1978; Uwajima et al. 1974). A solution of the recombinant enzyme (1.0 mg ml−1) showed an absorbance of 0.198 at 450 nm. In this solution, the molarity of the protein was estimated to be 19.0 mM based on the molecular mass (58 kDa) determined by SDS-PAGE. The molar adsorption coefficient of FAD (ɛ = 11.3 × 103 M−1 cm−1) was employed to calculate the concentration of FAD, 17.4 mM. This result indicating that the recombinant enzyme contained 1 mol of FAD per mole of protein was similar to that of the enzyme purified from strain DS-1 (Doukyu et al. 2008).
Physicochemical properties of the recombinant cholesterol oxidase
The enzyme was most active at pH 7.0 to 7.5 (Fig. 6a). The recombinant enzyme was stable from pH 3.0 to 11 after incubation for 1 h at 30°C (Fig. 6a). The optimum temperature at pH 7.0 was 65°C (Fig. 6b). Thermal stability was examined by incubating the enzyme in sodium phosphate (pH 7.0) buffer at various temperatures for 30 min (Fig. 6b). The enzyme was stable at temperatures from 4°C to 85°C. The enzyme retained about 79% and 27% of its activity after incubation for 30 min at 85°C and 90°C, respectively. However, the enzyme lost almost all activity after 30 min at 100°C.

Effects of pH and temperature on activity and stability of the recombinant cholesterol oxidase. a Effect of pH: enzyme activity (open circles) was assayed by following the consumption of oxygen at 30°C under the various pH conditions indicated in the figure. The pH stability (closed circles) was assayed after incubation at 30°C for 1 h under the various pH conditions indicated. The residual activity was examined by monitoring H2O2 generation at 30°C. The buffer systems (100 mM) used were glycine–HCl (pH 2.0–3.0), citrate–sodium citrate (pH 3.0–4.0), CH3COOH–CH3COONa (pH 5.0–5.5), NaH2PO4–Na2HPO4 (pH 5.5–7.5), Tris–HCl (pH 7.5–9.0), Na2CO3–NaHCO3 (pH 9.0–11.0), and NaCl–NaOH (pH 11.0–12.0). b Effect of temperature: enzyme activity (open circles) was assayed by following the formation of HCEO at pH 7.0 at the temperatures indicated in the figure. Thermal stability (closed circles) was assayed after incubation of the enzyme (0.2 U ml−1) dissolved in 100 mM phosphate buffer (pH 7.0) for 30 min at the temperatures indicated, and the relative activity was assayed by monitoring H2O2 generation at 30°C
We examined the effect of metal ions on the cholesterol oxidase activity by measuring enzyme activity at 30°C for 1 h in the presence of various metal ions. At a concentration of 1 mM, Ag+, Ca2+, Cu2+, Mg2+, Mn2+, Ni2+, and Fe2+ scarcely influenced the enzyme activity. However, the addition of 1 mM Zn2+ reduced the activity to 68% of that without a metal ion. A chelating agent, EDTA, did not show a significant inhibitory effect on the enzyme activity.
The enzyme was stable in the presence of 0.5% (wt vol−1) detergents, including Tween 20, Triton X-100, sodium cholate, sarcosyl, and Emal 20CM after 1 h at 45°C (Table 3). In the presence of SDS, the enzyme activity was only 12% of that without a detergent. The enzyme was stable also in the presence of 50% (vol vol−1) organic solvents, including dimethylsulfoxide, methanol, ethanol, isopropanol, ethyl acetate, butanol, chloroform, toluene, p-xylene, and cyclooctane after 24 h at 37°C. Treatment with acetone markedly inactivated the enzyme.
The pH and temperature activity and stability profiles, the activity in metal ions, and the stability in the presence of detergents and organic solvents of the recombinant enzyme were similar to those of native enzyme purified from strain DS-1 (Doukyu et al. 2008).
K m and V max values of cholesterol oxidases
We estimated the K m and V max values of the recombinant DS-1 enzyme and cholesterol oxidases from other bacterial origins from Lineweaver–Burk plots (Table 4) and found that the values were similar to those of the native enzyme purified from strain DS-1 (Doukyu et al. 2008). The K m value of the recombinant enzyme was lower than those of the enzymes from B. cepacia ST-200, P. fluorescens, and Streptomyces sp. SA-COO, but not those from Nocardia species. The V max value and V max/K m value (which was tentatively regarded as an enzyme efficiency) of the recombinant enzyme were the highest among the enzymes tested.
CD spectral analysis
The amino acid sequence of DS-1 cholesterol oxidase showed significant similarity to that of cholesterol oxidase from B. cepacia ST-200 (Table 1). The enzyme from B. cepacia ST-200 was stable from 4°C to 70°C but lost almost all activity after 30 min at 80°C, as reported previously (Doukyu et al. 2008). Therefore, the thermal stability of DS-1 cholesterol oxidase was higher than that of the enzyme from B. cepacia ST-200. The conformational transitions of the recombinant DS-1 enzyme and B. cepacia ST-200 enzyme were monitored by measuring the CD spectra at various temperatures (Fig. 7), and small changes of the CD spectra of DS-1 cholesterol oxidase were encountered at temperatures between 85°C and 90°C. Further increases in temperature had a significant impact on protein structure. The percentages of α-helix, β-sheet, turn, and random coil of DS-1 enzyme were as follows: at 60°C, 19.7%, 48.0%, 3.4%, and 28.9%; and at 95°C, 30.1%, 16.6%, 18.7%, and 34.6%, respectively. Thus, the increase of the α-helix, the turn, and the random coil and the decrease of the β-sheet were observed accompanying the conformational changes at high temperatures. Big changes of the CD spectra of B. cepacia ST-200 enzyme were observed at temperatures between 75°C and 80°C. The percentages of α-helix, β-sheet, turn, and random coil of B. cepacia ST-200 enzyme were as follows: at 60°C, 26.2%, 53.0%, 0%, and 20.8%; and at 80°C, 78.9%, 0% 0%, and 21.1%, respectively. In the case of B. cepacia ST-200 enzyme, the α-helix was significantly increased, and the β-sheet was completely lost at 80°C.

CD spectra of the recombinant cholesterol oxidase and B. cepacia ST-200 cholesterol oxidase at various temperatures. The CD spectra of cholesterol oxidases were measured between wavelengths of 195 nm and 250 nm. a The CD spectra of B. cepacia ST-200 cholesterol oxidase were measured at temperatures between 60°C and 85°C. b The CD spectra of DS-1 cholesterol oxidase were measured at temperatures between 60°C and 95°C
Discussion
There are two types of cholesterol oxidase that are different in their products (CEO or HCEO) from cholesterol (Doukyu and Aono 1999). CEO-forming cholesterol oxidase oxidizes cholesterol to CEO, with the consumption of 1 mol of O2 and the formation of 1 mol of H2O2 for every 1 mol of cholesterol oxidized. CEO-forming cholesterol oxidases have been reported from various microorganisms, including the genus Brevibacterium, Nocardia, and Streptomyces (Doukyu and Aono 1999; MacLachlan et al. 2000). On the other hand, HCEO-forming cholesterol oxidase oxidizes cholesterol to HCEO, with the consumption of 2 mol of O2 and the formation of 1 mol of H2O2 for every 1 mol of cholesterol oxidized. In a previous study, we showed that DS-1 cholesterol oxidase was the latter type of enzyme (Doukyu et al. 2008). HCEO-forming cholesterol oxidases have been reported from B. cepacia strain ST-200, Pseudomonas spp., and Chromobacterium sp. strain DS-1 (Doukyu and Aono 1999). Among these HCEO-forming enzymes, the cloning of a cholesterol oxidase gene was reported only from B. cepacia strain ST-200.
The sequence of a cholesterol oxidase gene from Chromobacterium has not been reported previously. Therefore, this is the first report of a gene encoding cholesterol oxidase from the genus Chromobacterium. Two forms of cholesterol oxidase have been identified in B. sterolicum, one containing the FAD cofactor non-covalently bound to the enzyme (BCO1) and another containing the cofactor covalently linked to the enzyme (BCO2; Croteau and Vrielink 1996). These two enzymes catalyze the same chemical reaction, although they have no significant sequence homology. BCO1 shows remarkable similarities (52% to 90%) to cholesterol oxidases or hypothetical proteins from actinomycetes, such as Rhodococcus and Streptomyces. These sequences contain a consensus sequence for FAD-binding, Gly-X-Gly-X-X-Gly, in the N-terminal region of the mature cholesterol oxidases (Croteau and Vrielink 1996; Ohta et al. 1991). The amino acid sequence of cholesterol oxidase from Chromobacterium sp. strain DS-1 did not show similarity to BCO1 or other proteins that have significant homology with BCO1, and it did not contain the consensus sequence for FAD-binding described above. By contrast, the sequence of DS-1 enzyme showed significant similarity (42% to 62%) to BCO2 and other cholesterol oxidases from R. erythropolis, S. ambofaciens, Burkholderia spp., and P. aeruginosa. The structure of BCO2 has been determined by X-ray crystallography and refined to high resolution (Coulombe et al. 2001). The structure suggested that the FAD cofactor was covalently bound to an active-site histidine (His121) via the C8α group of the flavin isoalloxazine ring. In addition, Glu475, located at the active-site cavity, was predicted to act as the base for both the oxidation and the isomerization steps of the catalytic reaction. Moreover, the structure indicated that the highly hydrophilic residues (Glu311, Glu432, Arg477, Glu551, Lys554, and Asn516) lining the cavity near the FAD cofactor probably play an important role in the reactivity of the cofactor. By a comparative amino acid sequence analysis, the amino acid residues His121, Glu475, Glu311, Arg477, Glu551, and Lys554 of BCO2 were conserved in the sequence of DS-1 enzyme as corresponding amino acid residues His107, Glu445, Glu299, Arg447, Glu518, and Lys524, respectively (Fig. 3). However, the amino acid residues Glu432 and Asn516 of BCO2 were replaced by corresponding amino acid residues of Ala402 and Asp486 in the sequence of DS-1 enzyme, respectively.
The amino acid sequence of cholesterol oxidase from strain DS-1 showed significant similarity to that of cholesterol oxidase from B. cepacia ST-200. The thermal stability of DS-1 cholesterol oxidase was higher than that of the enzyme from B. cepacia ST-200 (Doukyu et al. 2008). CD spectral analysis of the DS-1 enzyme and B. cepacia ST-200 enzyme at various temperatures showed that the conformational stabilities of these two enzymes were closely correlated with their respective thermal stabilities. As expected, the conformational stability of the DS-1 enzyme was higher than that of B. cepacia ST-200 enzyme at higher temperatures. In both enzymes, the transitions of the β-sheet to other secondary structures were observed at higher temperatures. Especially, the loss of the β-sheet of B. cepacia ST-200 enzyme was more distinctive than that of the DS-1 enzyme. The thermal stability of these enzymes might depend on the structural stability of the β-sheet at high temperatures.
Native cholesterol oxidase purified from strain DS-1 was more stable at high temperatures and in the presence of various organic solvents and detergents than were commercially available cholesterol oxidases (Doukyu et al. 2008). The recombinant enzyme of strain DS-1 also possessed these useful features for clinical applications and other reactions containing various organic solvents and detergents. In this study, we found that the catalytic efficiency of the recombinant enzyme from strain DS-1 was higher than those of the commercial enzymes. Moreover, we improved the production of the enzyme by recombinant E. coli, as compared with that by strain DS-1. The enzyme yield from the same culture volume was about 148-fold higher than that reported for strain DS-1 (Doukyu et al. 2008). The overproduction of the protein could allow its production on an industrial scale and shows its potential as a commercial enzyme.
References
Altschul S, Gissh W, Miller W, Mysers E, Lipman D (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Aono R, Doukyu N (1996) Stereospecific oxidation of 3β-hydroxysteroids by persolvent fermentation with Pseudomonas sp. ST-200. Biosci Biotechnol Biochem 60:1146–1151
Aono R, Doukyu N, Kobayashi H, Nakajima H, Horikoshi K (1994) Oxidative bioconversion of cholesterol by Pseudomonas sp. strain ST-200 in a water-organic solvent two-phase system. Appl Environ Microbiol 60:2518–2523
Biellmann J (2001) Resolution of alcohols by cholesterol oxidase from Rhodococcus erythropolis: lack of enantiospecificity for the steroids. Chirality 13:34–39
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cho HJ, Choi KP, Yamashita M (1995) Introduction and expression of the Streptomyces cholesterol oxidase gene (choA), a potent insecticidal protein active against Boll weevil larvae, into tobacco cells. Appl Microbiol 44:133–138
Coulombe R, Yue KQ, Ghisla S, Vrielink A (2001) Oxygen access to the active site of cholesterol oxidase through a narrow channel is gated by an Arg-Glu pair. J Biol Chem 276:30435–30441
Croteau N, Vrielink A (1996) Crystallization and preliminary X-ray analysis of cholesterol oxidase from Brevibacterium sterolicum containing covalently bound FAD. J Struct Biol 116:317–319
Dieth S, Tritsch D, Biellmann J-F (1995) Resolution of allylic alcohols by cholesterol oxidase isolated from Rhodococcus erythropolis. Tetrahedron Lett 36:2243–2246
Doukyu N, Aono R (1998) Purification of extracellular cholesterol oxidase with high activity in the presence of organic solvents from Pseudomonas sp. ST-200. Appl Environ Microbiol 64:1929–1932
Doukyu N, Aono R (1999) Two moles of O2 consumption and one mole of H2O2 formation with cholesterol peroxidation by cholesterol oxidase from Pseudomonas sp. ST-200. Biochem J 341:621–627
Doukyu N, Aono R (2001) Cloning, sequence analysis and expression of a gene encoding an organic solvent- and detergent-tolerant cholesterol oxidase of Burkholderia cepacia strain ST-200. Appl Microbiol Biotechnol 57:146–152
Doukyu N, Shibata K, Ogino H, Sagermann M (2008) Purification and characterization of Chromobacterium sp. DS-1 cholesterol oxidase with thermal, organic solvent, and detergent tolerance. Appl Microbiol Biotechnol 80:59–70
Fukuyama M, Miyake Y (1979) Purification and some properties of cholesterol oxidase from Schizophyllum commune with covalently bound flavin. J Biochem 85:1183–1193
Inouye Y, Tagnchi K, Fujii A, Ishimaru K, Nakamura S, Nomi R (1982) Purification and characterization of extracellular 3β-hydroxysteroid oxidase produced by Streptoverticillium cholesterolieum. Chem Pharm Bull 30:951–958
Isobe K, Shoji K, Nakanishi Y, Yokoe M, Wakao N (2003) Purification and some properties of cholesterol oxidase stable in detergents from gamma-proteobacterium Y-134. J Biosci Bioeng 95:257–263
Johnson TL, Somkuti GA (1991) Isolation of cholesterol oxidase from Rhodococcus equi ATCC33706. Biotechnol Appl Biochem 13:196–204
Kamei T, Takiguchi Y, Suzuki H, Matsuzaki M, Nakamura S (1978) Purification of 3β-hydroxysteroid oxidase of Streptomyces violascens origin by affinity chromatography on cholesterol. Chem Pharm Bull 26:2799–2804
Kazandjian RZ, Durdich JS, Kilbanov AM (1986) Enzymatic analysis in organic solvents. Biotechnol Bioeng 28:417–421
Laemmli UK (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 224:680–685
Lee S, Rhee H, Tae W, Shin J, Park B (1989) Purification and characterization of cholesterol oxidase from Pseudomonas sp. and taxonomic study of the strain. Appl Microbiol Biotechnol 31:542–546
Liu W-H, Meng M-H, Chen K-S (1988) Purification and some properties of cholesterol oxidases produced by an inducible and a constitutive mutant of Arthrobacter simplex. Agric Biol Chem 52:413–418
MacLachlan J, Wotherspoon ATL, Ansell RO, Brooks CJW (2000) Cholesterol oxidase: sources, physical properties and analytical applications. J Steroid Biochem Mol Biol 72:169–195
Ohta T, Fujishiro K, Yamaguchi K, Tamura Y, Aisaka K, Uwajima T, Hasegawa M (1991) Sequence of gene choB encoding cholesterol oxidase of Brevibacterium sterolicum: comparison with choA of Streptomyces sp. SA-COO. Gene 103:93–96
Pollegioni L, Gadda G, Ambrosius D, Ghisla S, Pilone M (1999) Cholesterol oxidase from Streptomyces hygroscopicus and Brevibacterium sterolicum: effect of surfactants and organic solvents on activity. Biotechnol Appl Biochem 30:27–33
Purcell JP, Greenplate JT, Jennings MG (1993) Cholesterol oxidase: a potent insecticidal protein active against Boll weevil larvae. Biochem Biophys Res Commun 196:1406–1413
Richmond W (1973) Preparation and properties of a cholesterol oxidase from Nocardia sp. and its application to the enzymatic assay of cholesterol in serum. Clin Chem 19:1350–1356
Sambrook J, Maniatis T, Fritsch E (1989) Molecular cloning: a laboratory manual. Cold Spring harbor Laboratory Press, Cold Spring Harbor, NY
Shirokane Y, Nakamura K, Mizusawa K (1977) Purification and some properties of an extracellular 3-hydroxysteroid oxidase produced by Corynebacterium cholesterolicum. J Ferment Technol 55:337–345
Smith AJ, Brooks CJW (1974) Application of cholesterol oxidase in the analysis of steroids. J Chromatog 101:373–378
Smith M, Zahnley J, Pfeifer D, Goff D (1993) Growth and cholesterol oxidation by Mycobacterium species in tween 80 medium. Appl Environ Microbiol 59:1425–1429
Srisawasdi P, Chaichanajarernkul U, Teerakranjana N, Kroll M (2008) Implementation of cellulomonas cholesterol oxidase for total serum cholesterol determination by the endpoint method. J Clin Lab Anal 22:50–58
Tomioka H, Kagawa M, Nakamura S (1976) Some enzymatic properties of 3β-hydroxysteroid oxidase produced by Streptomyces violascens. J Biochem 79:903–915
Uwajima T, Yagi H, Terada O (1974) Properties of crystalline 3β-hydroxysteroid oxidase of Brevibacterium sterolicum. Agric Biol Chem 38:1149–1156
Yang JT, Wu CS, Martinez HM (1986) Calculation of protein conformation from circular dichroism. Methods Enzymol 130:208–269
Acknowledgments
This work was supported in part by the Industrial Technology Research Grant Program in 2005 from New Energy and Industrial Technology Development Organization (NEDO) of Japan, the INOUE ENRYO Memorial Foundation for the Promotion of Sciences, and the Grant for the High Tech Research Center Program organized by the Ministry of Education, Culture, Sports, Science and Technology of Japan, since 2006. We thank Hiromi Kaneko and Kaori Yoshioka for their technical support. We thank Naoki Kajiyama, Kouzou Hirokawa, and Kazuki Shiga (Kikkoman Corp.) for their kind support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Doukyu, N., Shibata, K., Ogino, H. et al. Cloning, sequence analysis, and expression of a gene encoding Chromobacterium sp. DS-1 cholesterol oxidase. Appl Microbiol Biotechnol 82, 479–490 (2009). https://doi.org/10.1007/s00253-008-1775-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1775-9