
Abstract
The automated docking program DOCK 5.3.0 was applied to screening for quorum sensing inhibitors (QSIs) of Peudomonus aeruginosa from a database containing 51 active components of Traditional Chinese Medicines with antibacterial activity. Five potential QSIs were revealed by the computer-based virtual screening. The compounds 3, 4, 5, 6, 7 inhibit biofilm formation of P. aeruginosa at a concentration of 200 μM. Compound 4 (baicalein) does not inhibit the growth of P. aeruginosa; however, it significantly inhibits biofilm formation of the bacteria at a lower concentration of 20 μM and promoted proteolysis of the signal receptor TraR protein in Escherichia coli at 4–40 mM. Baicalein and ampicillin showed synergistic activity against P. aeruginosa. These results suggested that baicalein can interfere with quorum sensing system of P. aeruginosa and will be developed as antibacterial agent with novel target.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Traditional treatment of infectious diseases is based on compounds that kill or inhibit growth of bacteria. However, a major concern with this approach is the frequent development of resistance to antibiotics. Experience suggests that, as new drugs become widely deployed, resistance to these agents will emerge and spread as well. Successful control of antibiotic resistance will require both the continued development of new drugs and the judicious use of our current arsenal of antibiotics (Bergstrom et al. 2004).
Quorum sensing (QS) is a regulatory mechanism which enables bacteria to make collective decisions with respect to the expression of a specific set of genes. Gram-negative bacteria commonly use N-acyl homoserine lactones (AHLs) as QS signal molecules. The AHLs are synthesized by the family of LuxI homologue proteins. When the AHLs reach a certain threshold concentration, binding to receptor molecules (LuxR) is promoted, and the activated LuxR-AHL complex forms dimers or mutimers which, in turn, act as transcriptional regulator on target genes of the QS systems (Passador et al. 1993; Ochsner and Reiser 1995; Parsek and Greenberg 2000; Vaninni et al. 2002). The pathogenic bacterium Pseudomonas aeruginosa uses AHLs to coordinate the expression of a battery of virulence genes in a cascade of regulatory events (Passador et al. 1993). The QS systems of P. aeruginosa are also involved in elevating tolerance to antibiotic as well as tolerance to the activity of host immune systems (Anwar et al. 1990; Drenkard 2003). According to the Centers for Disease Control and Prevention, 65% of all infections in developed countries are caused by biofilms, which are bacterial communities that settle and proliferate on surfaces and are covered by an exopolymer matrix (Lewis 2007). It has been found that bacteria living in the biofilm mode of growth are often up to 1,000 times more tolerant to antibiotic than the planktonic cells (Anwar et al. 1990). This level will often exceed the highest deliverable doses and, thus, making efficient treatment impossible (Drenkard 2003). Considering the emergence of increasing antibiotic resistant bacteria, the use of signal-molecule-based drugs to attenuate bacterial pathogenicity rather than bacterial growth is attractive. The QS systems represent highly attractive targets for the development of novel therapeutics.
Some quorum sensing inhibitors (QSIs) such as halogenated furanone C30, patulin, penicillic acid, and garlic extract were found to make P. aeruginosa much more susceptible to tobramycin (Hentzer et al. 2003; Rasmussen et al. 2005a; Rasmussen et al. 2005b). A few chemical QSIs have been proven successful in the nematode and pulmonary mouse infection models; however, the compounds are unsuitable for human use. The halogenated furanones are unstable, the patulin and penicillic acid are mycotoxins, and the active components in garlic exist in very low concentrations; hence, they are not applicable to the treatment of human patients (Rasmussen and Givskov 2006).
As plants has co-existed with QS bacteria for millions of years, it can be expected that at least some of them produce QSIs in order to reduce the pathogenic capability of infective bacteria. Some plants have been used for thousands of years in Traditional Chinese Medicines (TCMs). Many components from TCMs have been identified as effective in the treatment of various inflammatory diseases such as gastritis, stomatitis, dermatitis, and pneumonia (Ma et al. 2005). Some QSIs should be screened from TCMs.
Discovery of inhibitors for a target protein can be greatly facilitated by the use of computer-aided drug design (CADD). In particular, virtual database screening approaches offer the potential to identify novel chemical entities with a high probability of binding to a target protein (Furci et al. 2007). The 3D structural information on QS transcriptional regulators from some Gram-negative bacteria (Agrobacterium tumefaciens, Escherichia coli, and P. aeruginosa) have been elucidated (Vannini et al. 2002; Yao et al. 2006; Bottomley et al. 2007). In the present study, by taking advantage of the 3D crystal structure of TraR (A. tumefaciens) combined with CADD approaches and biological assays, we have identified five novel inhibitors of TraR. In addition, one compound could eradicate the formation of P. aeruginosa biofilms, indicating its potential for development into novel anti-pathogenic drugs.
Materials and methods
Bacterial strains, plasmids, medium, and compounds preparation
P. aeruginosa PAO1 was cultured in Lubria-Bertani (LB) medium at 37°C. The traR gene (encoding TraR) was amplified from the plasmid pCF218 of A. tumefaciens WCF47(pCF372/pCF218; Zhu et al. 1998; provided by Dr. Winans of Cornell University) with primers TrarF (5-CGCGGATCCCATGCAGCACTGGCTG-3) and TrarR (5-CCGGAATTCTCAGATGAGTTTCCGC-3). The traR gene was further digested with BamHI and EcoRI and cloned into vector pET-17b (Novagen, Madison, WI, USA). The cloned plasmid (pETtrar) was transformed into E. coli strain Bl21(DE3). The LB broth containing 100 μg/mL ampicillin was used for growth and maintenance of E. coli strain Bl21(DE3)(pETtrar).
All the compounds (purity ≥ 98%) used in the study were purchased from Guangzhou Institute for Drug Control, China and dissolved in dimethyl sulphoxide (DMSO) except for compound 2 in ddH2O. Seven TCMs and their nine active components (Fig. 1) were listed in Table 1.

Chemical structures of compounds 1–9 and halogenated furanone C30. Compounds 1–9 were purchased from Guangzhou Institute for Drug Control
Ligands docking
The 3D structure of transcription activator protein TraR, obtained from Protein Data Bank (PDB ID code 1H0M), was used to dock. All the water molecules and B, C, D, E, F, G chains were removed; only A chain was used for the model. In the DOCK package, binding pocket was described by spheres. Each sphere touched the molecular surface at two points and had its radius along the surface normal of one of the points. The spheres were generated by the program sphgen which distributed as an accessory of DOCK. To define the binding pocket of chain A of TraR protein, N-(3-oxo-octanoyl)-L-homoserine lactone (3-oxo-C8-HSL; Fig. 2), the autoinducer of TraR protein, was used as ligand to select spheres. The spheres within 10 Å of ligand were selected. The sphere-selector module was used for the sphere selecting.

Schematic diagram of autoinducer 3-oxo-C8-HSL in chain A
The 51 compounds and halogenated furanone C30 were rigidly docked into the binding pocket. The box size was set as 6 Å, the grid space was 0.3 Å, and energy cutoff distance was 9,999 Å. Maximal orientations were set as 30,000. Default settings were used in Docking. Before docking, SYBYL program-package was used to add hydrogen atoms and charges to the protein (Tripos Associates 2001). All of the hydrogen atoms of protein were added and refined in the presence of explicit solvent with a progressive energy-minimization protocol using AMBER7 FF99 force field. The Termination Gradient is 0.01 kcal/mol, maximal iterations is 1,000 (Tripos Associates 2001). Docking studies were performed with the DOCK 5.3.0 program (Meng et al. 1993).
Effect on degradation of TraR in E. coli
E. coli Bl21(DE3)(pETtrar) was incubated in LB broth at 37°C with shaking. After the cell optimal density (OD600) reached 0.3, 400 μM Isopropyl-β-D-Thiogalactoside was added and continued to incubate for 4 h. Subsequently, 1.5 mL cultures were harvested by centrifugation (10,000×g) at 4°C for 2 min. The cells were washed twice with an equal volume of sterile distilled water. Then, the cells were mixed with 40 mM different compounds and 50 μL of sterile distilled water containing 100 μg/mL ampicillin and 100 μg/mL chlorampenicol. The mixtures were incubated at 30°C for 4 h and tested by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Effect on P. aeruginosa biofilm formation
The mass of biofilm was measured by the method described previously with modification (You et al. 2007). One milliliter of P. aeruginosa cells (OD600 = 1.0) were mixed with 200 μM different compounds and 1% (v/v) glycerol in 2 mL microfuge tubes, then, the tubes were incubated at 37°C for 3 days. The cultures in tubes were removed and the tubes were washed with sterile distilled water for three times to remove loosely associated bacteria. Subsequently, the tubes were air-dried. Each dried tube was stained with 1 mL 0.5% (w/v) crystal violet solution for 30 min and washed three times with sterile distilled water to remove excess crystal violet. The quantitative analysis of biofilm was performed by adding 1 mL 95% (v/v) ethanol into the tube for 15 min. Then, the crystal violet present in the ethanol solution was measured by spectrometry at 570 nm. To view the form of biofilm under microscope, cover glasses were added into 50 mL conical flasks containing bacteria, 1% (v/v) glycerol, and in the presence or absence (control) of 200 μM compound 4. After incubated at 37°C for 3 days, the dunked cover glasses were gently washed with sterile distilled water, Gram stained, and examined under microscope.
Effect on P. aeruginosa growth
To test for antibacterial activity of compound 4, 100 μL of P. aeruginosa (≈105 cfu/ml) was spread on LB plate and three filter paper discs were placed on the plate. Different solutions (20 μL of compound 4 (200 mM) plus 2 μL of ampicillin (1 mg/ml), 20 μL of DMSO plus 2 μL of ampicillin (1 mg/ml) and 20 μL of compound 4 (200 mM) singly) were added onto these discs, respectively. This plate was cultured at 37°C for 24 h and examined for growth inhibition zones.
Results
Virtual screening
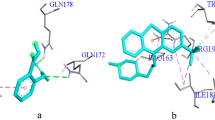
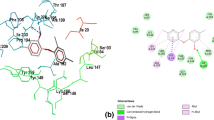
The virtual screening revealed a suite of new QSIs of A. tumefaciens. The analysis using Dock 5.3.0 program suggested that 20 compounds among 51 candidates got higher scores than halogenated furanone C30. These compounds might inhibit QS systems more efficiently than halogenated furanone C30. Compounds selected from the Dock based screening may be assumed to all structurally complement the target binding site; thus, they had potential to bind to the protein. We did not evaluate all 20 compounds experimentally. Eight available high-scoring and one low-scoring compounds were further selected for biological assay (Table 1). The predicted orientations of active compound 4 bound to TraR were generated with Chimera program (Pettersen et al. 2004) and shown in Fig. 3. Besides, more details about halogenated furanone C30 and compound 4 interacting with TraR residues were shown in Fig. 4.

a Map of TraR receptor and ligand crystal structure. b Superposition of ligand and compound 4. c Schematic presentation of interactions in TraR and compound 4

Possible binding modes of compounds 4 and halogenated furanone C30 to TraR protein region. a Interaction between TraR and compound 4. b Interaction between TraR and halogenated furanone C30. The energy score consists of van der Waals and electrostatic components, while the van der Waals are predominant. The energy score of compound 4 is higher than that of halogenated furanone C30, might be due to the phenyl group of compound 4 showed less complementarity with binding site of TraR protein
Effect of compounds on degradation of TraR protein in vivo
It is well known that intensity of the SDS-PAGE band correlates with the concentration of protein. Compared to the control, the concentration of TraR protein expressed in E. coli Bl21(DE3)(pETtrar) decreased in the presence of compound 4 (Fig. 5a). Further studies suggested that the concentration of TraR decreased as the concentration of compound 4 increased (Fig. 5b). The concentration of TraR quickly decreased (within 0.5 h) when 40 mM compound 4 was added.

Effect of compound 4 on TraR protein degradation in vivo. TraR protein was overproduced in E. coli, then, treated with different compounds and tested by SDS/PAGE. a TraR was treated with various compounds (40 mM) for 4 h: Cmpd 1–9 (compounds 1–9); b TraR was treated with various concentrations of compound 4 (0.4–40 mM) at different periods
Effect of compounds on in P. aeruginosa biofilm formation
Compounds 3, 4, 5, 6, 7 could inhibit biofilm formation of P. aeruginosa (Fig. 6a). Compound 4 showed the most potent inhibitory effect. Further studies showed that 20 μM compound 4 could significantly inhibit biofilm formation (P < 0.05, Fig. 6b). Most or all of the P. aeruginosa cells incubated with compound 4 (200 μM) detached from the surface and dispersed; however, the control cells adhered to and proliferated on the glass surface (Fig. 7). At a concentration of 200 mM, compound 4 singly did not inhibit the growth of P. aeruginosa, and clear inhibition zone around the disc was not observed (Fig. 8). The test bacteria were resistant to 2 μL of ampicillin (1 mg/ml). However, in the presence of compound 4, P. aeruginosa was significantly susceptible to ampicillin (Fig. 8). It demonstrated that compound 4 exhibited marked synergistic activity with ampicillin against P. aeruginosa.

Effect of different compounds on biofilm formation of P. aeruginosa. aP. aeruginosa was treated with various compounds (200 μM). b Effect of various concentrations (0–200 μM) of compound 4 on biofilm formation. All treatments were incubated at 37°C for 3 days and measured by spectrometry at 570 nm

Compound 4 inhibited biofilm formation of P. aeruginosa on cover glass. a Control. b The cover glass was not attached with biofilm in the presence of 200 μM of compound 4. Bar 10 μm

Synergistic activity of compound 4 and ampicillin against P. aeruginosa. a 20μL of compound 4 (200 mM) plus 2 μL of ampicillin (1 mg/ml). b 20μL of DMSO plus 2 μL of ampicillin (1 mg/ml). c 20 μL of compound 4 (200 mM)
Discussion
In pharmaceutical research, virtual screening has become a widely used approach and an integral part of many drug-discovery efforts. We applied such techniques in a search for novel QSIs. In this pharmacological class, there is a particular interest in finding new chemical entities due to the currently available QSIs are unsuitable for human use. Typically, virtual screening approaches involve database filtering to exclude compounds containing toxic, reactive, or otherwise undesired groups. In this study, a database of 51 compounds was screened because they are active components of TCMs with antibacterial activity.
Compounds 3, 4, 5, 6, 7 inhibited attachment of P. aeruginosa on glass surface at a concentration of 200 μM; however, only compound 4 promoted TraR proteolysis in E. coli. The results indicated that TCMs harbor numerous antibacterial components with different mechanisms. In the study, compound 4 (baicalein) could reduce concentration of TraR protein in E. coli. For the similarities in TraR turnover in E. coli and in A. tumefaciens (Zhu and Winans 2001), it is highly probable that this finding can also apply to A. tumefaciens. The result is consistent with that of halogenated furanones on LuxR protein (Manefield et al. 2002). P. aeruginosa biofilms are easily eradicated by tobramycin when they are pretreated with QSIs (Rasmussen and Givskov 2006). Baicalein-inhibited P. aeruginosa biofilm formation, however, did not affect its growth. So, baicalein probably penetrated the biofilms, interfered with cell signaling, and repressed QS in the majority of the biofilm cells. Moreover, baicalein-degrading TraR protein in vivo revealed the possible molecular mechanism by which baicalein interferes with QS system.
An effective and applicable QSI should require these properties (Rasmussen and Givskov 2006): (1) the QSI is a low-molecular-mass molecule the activity of which causes a significant reduction in the expression of QS-controlled genes, (2) the inhibitor exhibits a high degree of specificity for the QS regulator (i.e., the LuxR homolog) without side effects on either the bacteria or an eventual eukaryotic host, (3) the QSI should be chemically stable and resistant to metabolism and disposal by the higher host organism. Baicalein meets these requirements.
The dried roots of Scutellaria baicalensis (common name: Huangqin in China) have been widely employed for many centuries in TCMs as popular antibacterial and antiviral agents (Huang et al. 2005). They are effective against staphylococci, cholera, dysentery, pneumococci, and influenza virus. Baicalein, one of the major flavonoids contained in the dried roots, possesses a multitude of pharmacological activities (Huang et al. 2005). Remarkable synergies between baicalein and tetracycline and baicalein and β-lactams against methicillin-resistant Staphylococcus aureus have been reported (Fujita et al. 2005). Recently, synergism of the combination of baicalein and getamicin against vancomycin-resistant Enterococcus was also found (Chang et al. 2007). Furthermore, baicalein and ampicillin can act synergistically against P. aeruginosa in this study. These results indicated that targets of baicalein against bacteria are versatile. Extract of S. baicalensis is orally administered to patients. Thus, toxicity of baicalein is negligible, at least when administered orally.
The need to develop new anti-pathogenic drugs is especially acute in the case of P. aeruginosa infections in cystic fibrosis patients, where the natural antibiotic resistance of the bacteria and the ability to form biofilms account for significant mortality in such patients (VanDelden and Iglewski 1998). Therefore, baicalein described in the report provides the potential for a new class of antibacterial agents to target infections that are persistently difficult to combat with the current antibacterial agents.
References
Anwar H, Dasgupta MK, Costerton JW (1990) Testing the susceptibility of bacteria in biofilms to antibacterial agents. Antimicrob Agents Chemother 34:2043–2046
Bergstrom CT, Lo M, Lipsitch M (2004) Ecological theory suggests that antimicrobial cycling will not reduce antimicrobial resistance in hospitals. Proc Natl Acad Sci U S A 101:13285–13290
Bottomley MJ, Muraglia E, Bazzo R, Carfì A (2007) Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J Biol Chem 282:13592–13600
Chang PC, Li HY, Tang HJ, Liu JW, Wang JJ, Chuang YC (2007) In vitro synergy of baicalein and gentamicin against vancomycin-resistant Enterococcus. J Microbiol Immunol Infect 40:56–61
Drenkard E (2003) Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect 5:1213–1219
Fujita M, Shiota S, Kuroda T, Hatano T, Yoshida T, Mizushima T, Tsuchiya T (2005) Remarkable synergies between baicalein and tetracycline, and baicalein and beta-lactams against methicillin-resistant Staphylococcus aureus. Microbiol Immunol 49:391–396
Furci LM, Lopes P, Eakanunkul S, Zhong S, MacKerell AD Jr, Wilks A (2007) Inhibition of the bacterial heme oxygenases from Pseudomonas aeruginosa and Neisseria meningitidis: novel antimicrobial targets. J Med Chem 50:3804–3813
Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton JW, Molin S, Eberl L, Steinberg P, Kjelleberg S, Høiby N, Givskov M (2003) Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J 2:3803–3815
Huang Y, Tsang SY, Yao X, Chen ZY (2005) Biological properties of baicalein in cardiovascular system. Curr Drug Targets Cardiovasc Haematol Disord 5:177–184
Lewis K (2007) Persister cells, dormancy and infectious disease. Nature Rev Microbiol 5:48–56
Manefield M, Rasmussen TB, Henzter M, Andersen JB, Steinberg P, Kjelleberg S, Givskov M (2002) Halogenated furanones inhibit quorum sensing through accelerated LuxR turnover. Microbiology 148:1119–1127
Ma Z, Otsuyama K, Liu S, Abroun S, Ishikawa H, Tsuyama N, Obata M, Li FJ, Zheng X, Maki Y, Miyamoto K, Kawano MM (2005) Baicalein, a component of Scutellaria radix from Huang-Lian-Jie-Du-Tang (HLJDT), leads to suppression of proliferation and induction of apoptosis in human myeloma cells. Blood 105:3312–3318
Meng EC, Gschwend DA, Blaney JM, Kuntz ID (1993) Orientational sampling and rigid-body minimization in molecular docking. Proteins 17:266–278
Ochsner UA, Reiser J (1995) Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 92:6424–6428
Parsek MR, Greenberg EP (2000) Acyl-homoserine lactone quorum sensing in gram-negative bacteria: a signaling mechanism involved in associations with higher organisms. Proc Natl Acad Sci U S A 97:8789–8793
Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH (1993) Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260:1127–1130
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Rasmussen TB, Givskov M (2006) Quorum-sensing inhibitors as anti-pathogenic drugs. Int J Med Microbiol 296:149–161
Rasmussen TB, Bjarnsholt T, Skindersoe ME, Hentzer M, Kristoffersen P, Köte M, Nielsen J, Eberl L, Givskov M (2005a) Screening for quorum-sensing inhibitors (QSI) by use of a novel genetic system, the QSI selector. J Bacteriol 187:1799–1814
Rasmussen TB, Bjarnsholt T, Phipps RK, Christensen KB, Jensen PO, Andersen JB, Koch B, Larsen TO, Hentzer M, Eberl L, Hoiby N, Givskov M (2005b) Identity and effects of quorum-sensing inhibitors produced by Penicillium species. Microbiology 151:1325–1340
Tripos Associates (2001) SYBYL 6.9 [CP]. Tripos Associates, St. Louis
VanDelden C, Iglewski BH (1998) Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg Infect Dis 4:551–560
Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Marco SD (2002) The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J 21:4393–4401
Yao Y, Martinez-Yamout MA, Dickerson TJ, Brogan AP, Wright PE, Dyson HJ (2006) Structure of the Escherichia coli quorum sensing protein SdiA: activation of the folding switch by acyl homoserine lactones. J Mol Biol 355:262–273
You JL, Xue XL, Cao LX, Lu X, Wang J, Zhang LX, Zhou SN (2007) Inhibition of Vibrio biofilm formation by a marine actinomycete strain A66. Appl Microbiol Biotechnol 76:1137–1144
Zhu J, Winans SC (2001) The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc Natl Acad Sci U S A 98:1507–1512
Zhu J, Beaber JW, Moré MI, Fuqua C, Eberhard A, Winans SC (1998) Analogs of the autoinducer 3-oxooctanoyl-homoserine lactone strongly inhibit activity of the TraR protein of Agrobacterium tumefaciens. J Bacteriol 180:5398–5405
Acknowledgements
This work was supported by grants from the National High-Tech Development Program of China (2007AA091904, 2007AA09Z402) and National Natural Science Foundation (NO. 20231010). We gratefully thank the Open Laboratory for Marine Functional Genomics of State High-Tech Development Program, College of Life Sciences, Sun Yat-Sen University, Guangzhou 510275, P. R. China for providing the Sybyl 6.9 and thank the Kuntz Group for providing the DOCK program.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zeng, Z., Qian, L., Cao, L. et al. Virtual screening for novel quorum sensing inhibitors to eradicate biofilm formation of Pseudomonas aeruginosa . Appl Microbiol Biotechnol 79, 119–126 (2008). https://doi.org/10.1007/s00253-008-1406-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1406-5