
Abstract
Aspergillus nidulans PW1 produces an extracellular carboxylesterase activity that acts on several lipid esters when cultured in liquid media containing olive oil as a carbon source. The enzyme was purified by gel filtration and ion exchange chromatography. It has an apparent MW and pI of 37 kDa and 4.5, respectively. The enzyme efficiently hydrolyzed all assayed glycerides, but showed preference toward short- and medium-length chain fatty acid esters. Maximum activity was obtained at pH 8.5 at 40°C. The enzyme retained activity after incubation at pHs ranging from 8 to11 for 12 h at 37°C and 6 to 8 for 24 h at 37°C. It retained 80% of its activity after incubation at 30 to 70°C for 30 min and lost 50% of its activity after incubation for 15 min at 80°C. Noticeable activation of the enzyme is observed when Fe2+ ion is present at a concentration of 1 mM. Inhibition of the enzyme is observed in the presence of Cu2+, Fe3+, Hg2+, and Zn2+ ions. Even though the enzyme showed strong carboxylesterase activity, the deduced N-terminal amino acid sequence of the purified protein corresponded to the protease encoded by prtA gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
One of the largest groups of structurally related enzymes with diverse catalytic functions is the α/β-hydrolase fold family. This group serves as an example of divergent evolution, where proteins with different function have a common ancestral protein. Enzymes such as serine-proteases, lipases, esterases, and peptidases are members of this family. They have active site catalytic machinery composed of Ser, His, and Asp residues, which has evolved to efficiently act on substrates with different chemical compositions (Holmquist 2000). Interest in esterases, lipases, and serine-proteases has increased because they have several industrial applications in addition to their use in classic detergent formulations. One of these applications is the synthesis of fine chemicals achieved by catalysis in organic solvents to produce numerous high-value compounds (Sharma et al. 2001; Schmid and Verger 1998; Rao et al. 1998).
Thermostable enzymes are also of special interest for industrial applications because they are stable in typical operation conditions including high temperatures and extreme pH values. Thermostability can be obtained by immobilization of mesophilic enzymes or the use of thermophilic enzymes. Naturally thermostable enzymes are usually obtained from extremophilic microorganisms (Sinchaikul et al. 2001; Schmidt-Dannert et al. 1997). Nevertheless, some mesophilic bacteria and fungi produce thermostable enzymes that can be easily used, as these organisms can grow and produce enzymes in simple culture media and using soft culture conditions. This, is an advantage over extremophilic organisms. In any case, study of the basic biochemical properties of thermophilic enzymes from mesophiles and thermophiles may be important for potential applications of the biocatalysts in chemical and pharmaceutical industries (Sugihara et al. 1992; Hiol et al. 2000).
We have examined A. nidulans to identify potential carboxylesterases. In A. nidulans, several enzymes that hydrolyze ester bonds have been reported. Machado and de Castro-Prado (2001) reported differential esterase expression in this microorganism. Cutinolytic and lipolytic activities have been also detected (García-Lepe and Reyes 1997). Mayordomo et al. (2000) performed biochemical characterization of a lipase from strain WG 312. Ansari and Stevens (1983) found weak esterase activities in two proteinases isolated from strain BWB 272. The information in the available genome sequence database of A. nidulans (Aspergillus Sequencing Project, Whitehead Institute/MIT Center for Genome Research) and the database for the ESTs library constructed by Dr. Aramayo at Texas A&M University clearly show the presence of at least four different putative esterase genes, two subtilase genes, and many carboxylesterase genes. In the case of A. nidulans PW1, an enzymatic activity that generated clear halos on tributyrin agar seems to be fundamental for the organism survival as mutants that are unable to produce esterases could not be isolated by our research group after several mutagenic screening attempts (Kawasaki et al. 1995). We found only one carboxylesterase/lipase activity band on zymograms using trybutirin and o-nitrophenyl laurate from crude extracts induced with olive oil. The production of this enzyme increased 100-fold after optimization of the culture media using the same inducer (Peña-Montes, unpublished data). The present work describes the purification strategy used for this enzyme and its biochemical properties, which are significantly different from those of other reported strains. Unexpectedly, we found that the purified protein corresponded to the protease encoded by the previously reported prtA gene (Katz et al. 1994).
Materials and methods
Microorganisms and culture conditions
A. nidulans PW1 was donated by Dr. Jesus Aguirre, Institute of Cell Physiology, UNAM. The stock culture was maintained on silica gel slants at 4°C. Spores were produced on minimal nitrate agar plates as previously described (Kawasaki et al. 1995). The spores were collected in 0.1% NaCl solution and stored at 4°C. Sterile culture medium was inoculated with 1 × 106 spores/mL in 250-mL Erlenmeyer flasks containing 50 mL of culture media. The flasks were incubated at 37°C for 24 h on a rotatory shaker (200 rpm).
Culture medium
Minimal-optimized growth medium (adjusted to pH 6.5) was prepared as described by Kafer (1977) with the following modifications: starch (1.5%) as carbon source, yeast extract (0.5%), and olive oil (0.5%) as inducer.
Enzyme activity assays
1) Spectrophotometric assay. Esterase activity was quantified by a spectrophotometric method using o-nitrophenyl laurate (ONPL) or p-nitrophenyl laurate (PNPL) as substrate in accordance with Isobe et al. (1988). Esterase, 100 μL, was added to 900 μL of reaction buffer containing 50 mM potassium phosphate, pH 7.2, 1% Triton X-100, and 4 mg of ONPL or PNPL (Sigma). The reaction was carried out at 25°C and 1 U of enzyme activity was defined as the quantity of enzyme that released 1 μmol of o-nitrophenol per minute. Absorbance was continuously measured at 410 nm. When p-nitrophenyl esters with acyl group chain-lengths from C4–C14 were tested, we used a molar absorption coefficient of 14,500 l mol−1 cm−1 (410 nm, pH 7.5, 50 mM phosphate buffer) for p-nitrophenol.
2) Emulsion method. The method previously described by Sugihara and Shimada (1990) was used to determine lipolytic activity by titration of the free fatty acids released during hydrolysis of olive oil or tributyrin. A substrate emulsion was prepared with 5% of the substrate in a solution of 0.01% Tween 80, 50 mM of phosphate buffer pH 7.5, by emulsifying at maximum speed in a homogenizer (Ultraturrax T25, Janke and Kunkel). One milliliter of the enzyme was added to 4 mL of the emulsion. This mixture was incubated for 3 h at 37°C with constant agitation. The reaction was stopped by the addition of 10 mL of ethanol; the mixture was titrated to pH 10 with 50 mM KOH. The required volume of KOH is related to the fatty acids released by the esterase. One esterase unit is defined as the amount of enzyme releasing 1 μmol of fatty acid per minute.
Protein measurement
Protein concentrations were measured spectrophotometrically at 280 nm and by the Lowry (1951) method using bovine serum albumin as a standard.
Polyacrylamide gel electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 12.5% polyacrylamide slab gel was used to determine the purity and molecular weight of the enzyme by the method of Laemmli (1970). Low molecular weight markers (BioRad) were used as standards.
In situ esterase activity
After SDS-PAGE, esterase activity was detected by zymography. The acrylamide gel was washed by immersion in 0.1 M phosphate buffer, pH 7.5 for 30 min with constant agitation at room temperature (RT). A second wash was performed in the same conditions, except that the buffer contained 5% Triton X-100. Finally, a third wash in the same buffer with 0.5% Triton X-100 was performed. The acrylamide gel was then placed on a 1% agarose gel containing 2.5 mM o-nitrophenyl laurate (ONPL) and incubated at 37°C for 20 min.
When trybutirin was used as substrate, the acrylamide gel was placed on a 10% polyacrylamide gel containing 1% tributyrin and rhodamine B. The gels were incubated at 37°C for 20 min.
Esterase activity was also assayed on zymograms with α-naphthyl acetate. The protein samples were separated by SDS-PAGE and renatured as previously described. The gels were submerged for 5 min at RT in developing solution, 3 mM α-naphthyl acetate, and 5 min at RT in 1 mM Fast Red TR (Sigma). Both solutions contained 100 mM sodium phosphate buffer, pH 7.5. Esterase activity was detected by the appearance of purple-colored bands in the gels.
Isoelectric focusing
Electrofocusing was performed on a Phast System (BioRad) according to the instructions of the manufacturer. IEF 3–9 Phast Gels (Pharmacia) and high range (Bio-Rad) standards were used. Proteins were detected by Coomassie staining and zymography using ONPL as previously described.
Enzyme purification
After 24 h of growth, the culture broth was filtered to remove mycelia and was then lyophilized. After dissolving the lyophilized sample in 0.2 M Tris–HCl, pH 7.5, extraction with ethylic ether was performed using one volume of the sample and four volumes of ethylic ether. The sample was shaken vigorously and then incubated overnight at 4°C. The aqueous phase was collected and centrifuged at 15,000 rpm for 15 min at 4°C. The supernatant was assayed for activity and then applied to a column containing Ultrogel ACA-54 as support. The column was previously equilibrated with 0.2 M Tris–HCl pH 7.5. Column fractions that displayed activity were concentrated by ultrafiltration (Amicon, Beverly, MA). The concentrated enzyme solution was then fractionated by high-performance liquid chromatography (HPLC) using an ionic exchange column, Econo-Pack (Bio-Rad), pre-equilibrated with 0.1 M Tris buffer, pH 8. The protein was eluted with a linear gradient of NaCl from 0 to 2 M in the same buffer, with a flow rate of 0.8 mL min−1. The elution was fractionated, and each fraction was assayed for esterase activity. Fractions with esterase activity were pooled and concentrated by ultrafiltration. The purity and molecular mass of the purified enzyme were assessed by SDS-PAGE.
Effect of pH on esterase activity and stability
To determine the optimal pH for esterase activity, the enzymatic rate was assayed using different pH values ranging from 5 to 10. The buffers used for pH values of 5, 6, 7–8.5, and 9–10 were acetate, MES, Tris, and CHES, respectively. The effect of the pH on enzyme stability was tested using the same buffer solutions. The samples were incubated without substrate for 12 and 24 h at 37 and 4°C. After incubation, the pH values remained constant, and the remaining activity was evaluated by the spectrophotometric method.
Effect of temperature on esterase activity and stability
To study the effect of temperature, enzymatic activity was measured for 15 min at 30, 40, 50, 60, 70, and 80°C at pH 8.5, with the relevant controls. Thermostability of the esterase was investigated by measuring the remaining activity at 25°C after incubation of the enzyme without substrate in 0.1 M Tris–HCl, pH 8.5, at various temperatures for 15, 30, and 60 min.
Effect of metal ions, EDTA, and SDS on esterase activity
The effect of metal ions and SDS on esterase activity was measured using the spectrophotometric assay. The reaction mixture was supplemented with various cations, ethylenediaminetetraacetic acid (EDTA), or SDS at a final concentration of 1 or 10 mM. The cations were Ba2+, Ca2+, Cu2+, Co2+, Fe2+, Zn2+, Fe3+, Hg2+, and Mn2+.
Effect of detergents on esterase stability
The enzyme solution was incubated for 1 h at 30°C in 0.1 M Tris buffer, pH 8, containing 0.1 or 1% (w/v) of each detergent. Esterase activity was assayed by the spectrophotometric method and compared with the activity of the enzyme in the absence of detergent. The detergents used were Span-80, Tween-80, Triton X-100, and taurocholic acid.
Substrate specificity
Substrate specificity for different mono-, di-, and triacylglycerols was determined using the emulsion method. A substrate emulsion was prepared by emulsifying triacylglycerols (80 μM), diacylglycerols (40 μM), or monoacylglycerols (40 μM) in 0.1 M Tris buffer, pH 8. One hundred microliters of the enzyme solution was added to 3 mL of the emulsion, and this mixture was incubated for 3 h at 37°C under constant agitation. Lipolytic activity was measured by titration of the fatty acids with 50 mM KOH.
Stability in organic solvents
The enzyme solution was incubated under constant agitation for 1 h at 30°C in 0.1 M Tris buffer, pH 8, containing 15, 30, or 50% (v/v) of each solvent. Esterase activity was tested by the spectrophotometric assay and compared with the lipolytic activity of the enzyme in the absence of organic solvent. The tested solvents were ethanol, methanol, acetone, DMSO, isopropyl alcohol, and isopropyl ether.
Amino acid sequencing and identification
After SDS-PAGE, the protein was electroblotted on a polyvinylidenedifluoride membrane (Immobilon, Bio-Rad) by the Towbin method (Towbin et al. 1979) for further sequencing. The N-terminal sequence was determined by automated Edman degradation (Edman 1950) on a gas-phase protein sequencer (LF 3000, Beckman Instruments,) equipped with an online HPLC system (Beckman System Gold, Beckman Instruments). HPLC equipment included a diode array detector with settings at 268 and 293 nm for signal and reference, respectively. A Beckman Spherogel Micro PTH (2×150 mm) column, and standard Beckman sequencing reagents were used for analysis.
Internal protein sequencing and identification
Digest of the protein with trypsin was done in situ in the gel. Peptides were extracted from the gel matrix and analyzed by peptide mass fingerprinting and peptide sequencing with a matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI TOF MS) in reflector mode DHBS as matrix. Tandem mass (MS/MS) spectra of two peptides were recorded by a MALDI TOF/TOF (4700 Proteomics Analyzer).
Results
Esterase activity of the crude extract on zymograms
A crude extract of cells grown for 24 h in optimized medium after induction with olive oil was used to assess the hydrolysis of trybutirin and ONPL on zymograms. In both cases, we found only one band of activity corresponding to a molecular weight of 37 kDa (data not shown). The crude extract was used to test for the hydrolysis of short acyl-chain aromatic esters such as α-naphthyl acetate. We found three band activities on this zymogram. One weak band corresponded to a molecular weight of 37 kDa, and the two strong bands had molecular weights of 29 and 22 kDa (Fig. 1).

Zymogram for esterase activity. Proteins were separated by SDS-PAGE with a 12.5% gel. Lanes 1–2 (protein coomassie brilliant blue stains) and lanes 3–4 (esterase activity stains with α-NA), were crude extract after 24h of growth. Lane 1: low molecular weight marker proteins
Enzyme purification
A. nidulans PW1 was cultivated in optimized medium for 24 h. Purification factors and recoveries at each step are summarized in Table 1. The enzyme was purified 49.73-fold with a yield of 49.67% from the crude extract. After the purification procedure, fractions containing the protein exhibited activity in the form of a single band on SDS-PAGE by Coomassie blue and silver staining with an apparent molecular mass of approximately 37 kDa (Fig. 2). Electrofocusing assays followed by zymography with ONPL showed that the isoelectric point of the enzyme is 4.5.

SDS-PAGE electrophoresis gel of purified Aspergillus nidulans carboxylesterase. a) Protein silver stains. Lane 1: low molecular weight marker proteins and lanes 2–3: Purified protein. b) Protein Coomassie brilliant blue stains. Lanes 4–6: Purified protein and lane 8: low molecular weight marker proteins
Effect of pH and temperature on carboxylesterase activity and stability
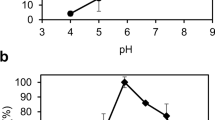
The enzyme was active in the pH range of 7 to 10, and the highest activity was found at pH 8.5 (Fig. 3). This value was the same for the activity in the crude extract and the pure enzyme preparation. Purified enzyme was found to be more stable at pH ranges from 8–11 for 12 h at 37°C. After 24 h, the enzyme was found to be more stable at pH ranges of 6–10. On the other hand, at 4°C for 12 h, its stability was better at a pH range of 7 to 11. After 24 h of incubation in the same temperature, the enzyme was more stable at pH range of 8–11 (data not shown).

Dependence of enzyme activity with pH for pure enzyme (○) and crude extract (●). Assay was carried out by spectrophotometric method using 0.1 M of the following buffers: acetate, pH 5; MES, pH 6; phosphate, pH 7; Tris-HCl, pH 8 and 8.5; CHES, pH 9 and 10. Obtained values are average of 3 measurements
The purified enzyme and the crude extract were more active in temperature ranges of 30 to 60°C, showing a maximum activity at 40°C (Fig. 4). After incubation for 15 or 30 min, the enzyme was stable from 30 to 70°C, retaining more than 80% of its initial activity. When the temperature was elevated to 80°C for 15 min, the enzyme lost 50% of its initial activity. After 30 min of incubation at 80°C, the activity dropped by as much as 80%. The enzyme remained stable for more than 30 min when it was incubated at 40°C, which is the optimal temperature for activity in our assay conditions.

Dependence of enzyme activity with temperature for pure enzyme (○) and crude extract (●). Inset: Arrhenius plot for enzyme inactivation. Effects were determined by spectrophotometric method using 50 mM phosphate buffer, pH 7.5. Obtained values are average of 3 measurements
Our study of the inactivation rate of pure enzyme is shown in the inset of Fig. 4. The enzyme is relatively stable below 60°C, while the inactivation rate increases dramatically above this temperature. This behavior is normal as the temperature increases the rate of the inactivation process. The Arrhenius plot for the inactivation process seems to be linear, which allows for calculation of the Arrhenius energy of activation, E a, for thermal-enzyme inactivation, 65.96 kJ mol−1.
Effect of metal ions and inhibitors on esterase activity
The presence of 1 mM of almost all tested metals and inhibitors slightly affected activity. Notably, Cu2+ reduced the esterase activity by 65% and, unexpectedly, Co2+ and Fe2+ increased activity up to 156 and 238%, respectively. When the concentration of metals and inhibitors was increased to 10 mM, the detrimental effects of Cu2+, Fe3+, Fe2+, Hg2+, and Zn2+ were more evident. In the same conditions, the chelating agent EDTA did not affect the esterase activity, suggesting that it is not a metalloenzyme. The denaturing effect of SDS was also noticeable at 10 mM (Fig. 5).

Effect of different ions, EDTA and SDS in activity. 1 mM (light gray) and 10 mM (gray). Activities are expressed relative to activity of enzyme in absence of tested agents. Obtained values are average of 3 measurements
Effect of detergents on esterase activity
The addition of 0.1% (w/v) detergent did not affect esterase activity with the exception of Tween-80, which increased activity by 160%. In the presence of 1% detergent, Tween-80 increased activity by 126%, while taurocholic acid slightly increased activity at 0.1%. Triton X-100 and Spa- 80 decreased activity at both concentrations (Table 2).
Substrate specificity
The purified esterase was able to hydrolyze synthetic substrates with acyl group chain-lengths of C4, C12, and C14 and displayed higher activity (85% higher) toward C4 chain-lengths (p-nitrophenyl butyrate; data not shown). The enzyme was more active on triglycerides with short chains (C4 and C6) than on mono- or diglycerides such as monocaprin, monopalmitoin, monoolein, dicaprin, and diolein. However, the enzyme showed higher activity toward long-chain mono- or diglycerides than toward long-chain triglycerides (C10 and C18; Fig. 6).

Substrate preference toward triacylglicerides (dark gray), diacylglycerides (gray) and monoacylglycerides (light gray) of different chain length for pure enzyme. Activities are expressed relative to activity on tributyrin. Carboxylesterase activity was determined by emulsion method using 80 μM of each triglyceride and 40 μM of each diacyl and mono glyceride in 50mM Tris-HCl pH 8 at 37°C. Obtained values are average of 3 measurements
Effect of organic solvents on esterase stability
The pure enzyme maintains excellent stability in all organic solvents assayed at a concentration of 15% for 1 h (Fig. 7). Moreover, isopropyl ether increased the esterase activity by about 60%. Higher concentrations (30%) of methanol and ethanol increased the esterase activity by 10 and 20%, respectively, whereas acetone and dimethylsulfoxide (DMSO) decreased the activity by 40%. Higher concentrations (50%) resulted in a severe reduction of activity except for isopropyl ether, which produced a 10% increase in esterase activity.

Effect of different organic solvents on enzyme stability at 15 (light gray), 30 (gray) and 50% (dark gray) (v/v). EA, ethyl alcohol; MA, methyl alcohol; DMSO, dimethyl sulfoxide; IPA, isopropyl alcohol; IPE, isopropyl ether; AC, acetone. Obtained values are average of 3 measurements
Protein sequencing and identification
The following is the N-terminal sequence: ALTSQSGAPW (Fig. 8). The protein was also digested with trypsin. Two prominent signals in the mass spectrum (MH+ 2801 and MH+ 3804) were observed. The resulting peptides were extracted and analyzed by amino acid sequencing One of the peptides was near the N-terminal region (peptide 1), and the other corresponded to the C terminus (peptide 2; Fig. 8). The resulting peptide sequences were used to search the A. nidulans genome database. The N-terminal amino sequence and both peptides identified the protein as AN5558.1. This protein corresponds to the alkaline protease encoded by the prtA gene (Katz et al. 1994).

Comparison of resulting N-terminal, peptide 1 and peptide 2 sequences with complete sequence of identified alkaline protease (AN5558.1) obtained from A. nidulans genome database. N-terminal, peptide 1 and peptide 2 sequences are marked by bold upper cases. Peptide 2 is the C-terminal peptide in the protein
Discussion
In spite of the high number of esterase and lipase genes in the Aspergillus genome (Aspergillus Sequencing Project, Whitehead Institute/MIT Center for Genome Research), results obtained in this work indicate that only three relevant proteins were present in the crude extract after induction with olive oil. These proteins were observed as three bands with esterolytic activity using alpha naphtyl acetate and one band using tributyrin, ONPL, or PNFL.
After the purification process, a single band of 37 kDa with a pI of 4.5 was obtained. Unexpectedly, the amino acid sequence of the N- and C-terminal regions of this esterolytic protein corresponded to the reported hypothetical prtA gene product, an alkaline protease (Katz et al. 1994). Comparison of alkaline protease gene sequences in the genus Aspergillus (including the prtA gene) to other alkaline protease enzymes indicated that they all belonged to the subtilase enzyme family (Siezen et al. 1991). We compared amino acid sequences near the amino acids that compose the putative catalytic triad, and we found that these regions in alkaline proteases from Aspergillus genus are conserved and are similar to the sequences of subtilisin and thermitase (Fig. 9).

Comparison of conserved regions and putative catalytic amino acids of Aspergillus nidulans serin protease with those of serin proteases of subtilase family, reported from other microorganisms. (A. flav, Aspergillus flavus elastinolytic serine protease; A. fum, A. fumigatus alkaline protease; A. ory, A. oryzae alkaline protease; P. chry, Penicillium chrysogenum alkaline serin protease; Prot B, protease B of Saccharomyces cerevisiae; Subtilisin, Bacillus amyloliquifaciens subtilisin; Thermitase, Thermoactinomyces vulgaris alkaline protease). Highlighted amino acids represent the putative catalytic triad
The amino acid sequence of the protein encoded by the prtA gene is shown in Fig. 8. The protein is 403 amino acids, which correspond to a molecular mass of 42 kDa. The deduced N-terminal sequence (ALTSQSGAPW) matched a.a. 120–128. The majority of subtilases are synthesized as precursors with a pre- and pro-sequence extension at the N-terminus of the mature protein (Bryan et al. 1995). At the amino terminal region of the complete protein, a series of uncharged amino acid residues with a high content of hydrophobic amino acids were found. Based on signal sequence cleavage prediction (Bendtsen et al. 2004), the most likely cleavage site in the present protein is between Gly-20 and Ala-21. This signal sequence is expected to direct transport of the nascent polypeptide chain across the ER membrane into the ER lumen (Hattori et al. 1987). Ninety-nine amino acids precede the deduced N-terminal sequence. The rather long propeptide functions as a protease self-inhibitor and an intramolecular chaperone that guides correct folding of the mature protein (Yabuta et al. 2001, 2003; Fig. 8). The mature alkaline serine protease has a calculated molecular mass of 28.6 kDa; however, our experiments indicate that the purified protein has an apparent molecular mass of 37 kDa. This shift in the molecular weight could be the result of posttranslational processing of the protein.
As we were looking for a carboxylesterase, the assays performed in this work were designed to evaluate the carboxylesterase activity of this protein. Its properties as a protease remain to be studied. Our preliminary results indicate that the enzyme can hydrolyze elastin congo red and hide power azure (data not shown).
Many activity assays reported in this paper were performed with soluble esters such as PNPB, PNPL, and PNPP, demonstrating that these non-interface-forming substrates can be hydrolyzed by this enzyme. This provides an insight into its esterase behavior. Nevertheless, the enzyme was also able to weakly hydrolyze insoluble triacylglycerols. Previous work by our group (Peña-Montes, unpublished data) demonstrated that the esterase of A. nidulans PW1 does not present interfacial activation. Enzymes are flexible structures that interact with different substrates with different hydrolysis rates. However, the real impact of enzyme activity for organisms is that their multiplicity of substrates fulfill the biological role intended for the enzyme, whereas the role of an enzyme in biotechnology is catalysis of the desired reactions with minimal or no side products. Regardless of the nature of ester hydrolysis catalyzed by this enzyme, it is clear from our data that it has greater affinity toward short-chain fatty acids for all tested triglycerides and p-nitrophenyl derivatives. The same behavior was observed for diglycerides and monoglycerides. The presence of unsaturated bond at C1, C2, or C3 produced interesting changes in catalytic activity, effecting a greater change when the unsaturated bond is at position 2 compared to position 3. This effect may be related to the ability to form an enzyme–substrate complex, as this position is very close to the ester group that binds to the active site pocket.
The selectivity toward different substrates observed for this enzyme is one of the more attractive properties of subtilases. It has been reported that subtilisins are hydrolytic enzymes with multifunctional properties such as broad substrate specificity and regiospecificity (Lo et al. 2003; Chang et al. 1995). Even though their more well-known substrates are peptides, they can weakly hydrolyze a wide variety of ester bonds, including short chain and aromatic fatty acids (Liu and Tamp 2001; Murayama et al. 2002; Sroga and Dordick 2001; Klibanov 2001). The biochemical properties of the enzyme include higher activity at pH 8.5 and 40°C in our assay conditions. Previous reports of fungal esterases suggest that these enzymes are usually more active at pH values near neutrality or slight acidity. A carboxylesterase from A. nidulans WG312 has higher activity at pH 6.5 and 40°C (Mayordomo et al. 2000). The protease from A. nidulans PW1 displayed alkaliphilic behavior. Acidic and neutral proteases from A. nidulans have been characterized, but there is little information about alkaline proteases in this microorganism (Ansari and Stevens 1983; Cohen 1973a, b).
The results shown in this work indicate that the protease from A. nidulans PW1 is very sensitive to pH inactivation. This is revealed by inactivation rates at different pH values (data not shown). This enzyme demonstrates maximum stability at pH 8. At this pH, it retains 99% of its activity after a 12-h incubation period. This is a considerably long incubation time for a free enzyme. The inactivation rates increase as the H+ concentration increases or decreases, suggesting an important role for this ion in the inactivation of this enzyme.
The observed inactivation rates at different temperatures indicate very high stability from 30 to 60°C for 24 h. The inactivation rate is dramatically different at higher temperatures (Fig. 4). The Arrhenius energy of activation, E a, value for this enzyme is 65.96 kJ mol−1 (inset of Fig. 4), which is small compared to other enzymes (150–400 kJ mol−1; Gordon et al. 2002; Fachin et al. 2002). E a strongly depends on the assay conditions, such as pH, ionic strength, and the nature and concentration of other molecules in the reaction medium. The E a value is related to the sensitivity of a process to temperature changes and depends on the free energy change, ΔG°. For an inactivation process with a very negative ΔG°, the E a should be high to maintain stability of the active form (Cornish-Bowden 1995). It must be demonstrated that the ΔG° energy required for the inactivation process of this enzyme is not very negative according to its E a value and allows thermostability. Further research on the thermal inactivation of this enzyme is necessary to clarify this point.
Some enzymes bind metal ions such as Ca2+, which changes their catalytic properties. This has been described for A. terreus, Botryits cinerea, A. niger, and Humicola lanuginosa lipases (Raman et al. 1998; Höfelmann and Hartmann 1985; Comménil and Belingheri 1995). This ion does not significantly alter the activity of the assayed esterase. Surprisingly, 1 mM Fe2+, which usually inhibits esterase activity (Ibrahim and Hayashi 1987; Kohno and Kugimiya 1994; Kundu and Basu 1987; Mase et al. 1995; Asahara and Matori 1993), increases the enzymatic activity in our assays. However, when the concentration of Fe2+ is raised to 10 mM, an inhibitory effect becomes apparent. This behavior may be related to multiple Fe2+ binding sites on the protein with different affinities, such that occupancy of a few sites results in activation of the enzyme, whereas occupancy of more sites distorts the structure and inhibits the enzymatic activity of the protein. The enzymatic activity was also tested in the presence of Fe2+ supplemented with EDTA, which reduces the enhancing effect exerted by Fe2+ alone (data no shown). Further research is needed to elucidatee of the nature of this activation–inhibition effect. The ions Cu2+, Fe3+, Hg2+, and Zn2+ demonstrated their commonly observed inhibitory effect on esterase activity. However, Cu2+ ion displayed a more dramatic effect, reducing activity to 10% (Fig. 5). These data strongly suggest the presence of several binding sites within the protein that alter its structure upon metal-ion binding, which may account for the catalytic changes observed. In contrast, the addition of the metal chelator EDTA did not produce any significant change in the activity, suggesting thus that the enzyme does not require any metal ion to carry out its catalytic function. To confirm that purified protein is a serine protease, an inhibitory experiment using phenylmethylsulphonyl fluoride (PMSF) 10 mM was performed. PMSF did inhibit enzyme activity, confirming its serine protease nature (data no shown).
Surfactants or detergents are usually recognized as chaotropic agents that in high concentrations, may produce protein denaturation by means of their interaction with hydrophobic and hydrophilic regions of the proteins. Nevertheless, surfactants are routinely incorporated into protein formulations to solubilize hydrophobic proteins or to act as chaotropic agents to prevent protein aggregation. The esterase studied in this work was not sensitive to the addition of 1 mM SDS, although activity was completely lost when the concentration was raised to 10 mM. Interestingly, Tween 80 (polyoxyethylenesorbitane monooleate) increased the esterase activity up to 160%, in contrast to the complete inhibition observed for esterases of Bacillus thermocatenulatus (Schmidt-Dannert and Schmid 1996) and Penicillium expansum (Stocklein et al. 1993). The nature of this activation is unclear, but it is obvious that Tween 80 is the largest of the molecules tested and has the largest hydrophobic surface. Surfactants not only facilitate oil drop dispersion, but can also bind to proteins and induce structural changes that for Tween 80, appear to be stimulatory.
Esterase activity in organic solvents has been intensively studied as these catalytic systems generate high-value products. Our studies of the stability of the protease of A. nidulans PW1 in organic solvents suggest that enzymatic behavior is differentially affected by various solvents. We found that the solvent that best conserves its hydrolytic properties is isopropyl ether. Nevertheless, further research on synthesis reactions in organic solvents is necessary.
It has been shown that the enzyme studied by our group is different from those previously described in A. nidulans. This enzyme could be an interesting structural model for metal–protein interactions as can be envisaged from the activation–inhibition patterns observed for the metal ions investigated in this work. Its thermostability and optimal temperature and pH make this enzyme an interesting model for industrial applications. It is important to develop a recombinant system to produce this enzyme. These research efforts are necessary to obtain more information about alkaline proteases in A. nidulans for potential industrial applications.
References
Ansari H, Stevens L (1983) Purification and properties of two neutral proteinases from Aspergillus nidulans. J Gen Microbiol 129(6):1637–1644
Asahara T, Matori M (1993) Production of two types of esterases with opposite positional specificity by Geotrichum sp. Biosci Biotechnol Biochem 57:390–394
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795
Bryan P, Wang L, Hoskins J, Ruvinov S, Strausberg S (1995) Catalysis of a protein folding reaction: mechanistic implications of the 2–0 A structure of the subtilisin–prodomain complex. Biochemistry 34:10310–10318
Chang RC, Chen JC, Shaw JF (1995) Vibrio mimicus arylesterase has thioesterase and chymotrypsin-like activity. Biochem Biophys Res Commun 213(2):475–483
Cohen BL (1973a) The neutral and alkaline proteases of Aspergillus nidulans. J Gen Microbiol 77(2):521–528
Cohen BL (1973b) Regulation of intracellular and extracellular neutral and alkaline proteases in Aspergillus nidulans. J Gen Microbiol 79:311–320
Comménil P, Belingheri L (1995) Purification and properties of an extracellular esterase from the fungus Botrytis cinerea. Lipids 80:351–356
Cornish-Bowden A (1995) Fundamentals of enzyme kinetics. Portland, London
Edman P (1950) Method for the determination of the amino acid sequence in peptides. Acta Chem Scan 4:289–298
Fachin D, Van Loey A, Indrawati L, Hendrick M (2002) Thermal and high-pressure inactivation of tomato polygalacturonase: a kinetic study. J Food Sci 67:1610–1615
García-Lepe RO, Reyes F (1997) Esterases in autolysed cultures of filamentous fungi. Lett Appl Microbiol 25:127–130
Gordon EA, Sekine Y, Watanabe N, Barret DM (2002) Thermal inactivation of pectin methylesterase, polygalacturonase and peroxidase in tomato juice. J Agric Food Chem 50:6153–6159
Hattori T, Ichihara S, Nakamura K (1987) Processing of a plant vacuolar protein precursor in vitro. Eur J Biochem 166(3):533–538
Hiol A, Jonzo M, Rugani N (2000) Purification and characterization of an extracellular esterase from a thermophilic Rhizopus oryzae strain isolated from a palm fruit. Enzyme Microb Technol 26:421–430
Höfelmann M, Hartmann J (1985) Isolation, purification and characterization of esterase isoenzymes from a technical Aspergillus niger. J Food Sci 50:1721–1725
Holmquist M (2000) Alpha/beta-hydrolase fold enzymes: structures, functions and mechanisms. Curr Protein Pept Sci 1(2):209–235
Ibrahim CO, Hayashi M (1987) Purification and some properties of a thermostable esterase from Humicola lanuginosa. Agric Biol Chem 51:37–45
Isobe K, Akiba T, Yamaguchi S (1988) Crystallization and characterization of esterase from Penicillium cyclopium. Agric Biol Chem 52:41–47
Kafer E (1977) Meiotic and mitotic recombination in Aspergillus and its chromosomal aberrations. Adv Genet 19:33–131
Katz ME, Rice RN, Cheetham BF (1994) Isolation and characterization of an Aspergillus nidulans gene encoding an alkaline protease. Gene 150:287–292
Kawasaki L, Farres A, Aguirre J (1995) Aspergillus nidulans mutants affected in acetate metabolism isolated as lipid nonutilizers. Exp Mycol 19(1):81–85
Klibanov AM (2001) Improving enzymes by using them in organic solvents. Nature 409:241–246
Kohno M, Kugimiya W (1994) Purification, characterization and crystallization of two types of esterases from Rhizopus niveus. Biosci Biotech Biochem 58:1007–1012
Kundu M, Basu J (1987) Isolation and characterization of an extracellular esterase from the conidia of Neurospora crassa. J Gen Microbiol 133:149–153
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Liu CF, Tamp JP (2001) Subtilisin-catalyzed synthesis of amino acid and peptide esters. Application in a two-step enzymatic ligation strategy. Org Lett 3(26):4157–4159
Lo YC, Lin SC, Shaw JF, Liaw YC (2003) Crystal structure of Escherichia coli thioesterase I/protease I/lysophospholipase L1: consensus sequence blocks constitute the catalytic center of SGNH-hydrolases through a conserved hydrogen bond network. J Mol Biol 330(3):539–551
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Machado MF, de Castro-Prado MA (2001) Differential esterase expression in developmental mutants of Aspergillus nidulans. Biochem Genet 39:357–368
Mase T, Matsumiya Y, Akiba T (1995) Purification and characterization of a new esterase from Fusarium sp. YM-30. Biosci Biotechnol Biochem 59:1771–1772
Mayordomo I, Randez-Gil F, Prieto J (2000) Isolation, purification and characterization of a cold-active esterase from Aspergillus nidulans. J Agric Food Chem 48:105–109
Murayama T, Nagasawa S, Goto M (2002) Enzymatic synthesis of sugar amino acid esters in organic solvents. J Biosci Bioeng 94(4):357–361
Raman P, Rajendra K, Rani G (1998) Purification and characterization of a regiospecific esterase from Aspergillus terreus. Biotechnol Appl Biochem 28:243–249
Rao MB, Tanksale AM, Ghatge MS, Deshpande VV (1998) Molecular and biotechnological aspects of microbial proteases. Microbiol Mol Biol Rev 62(3):597–635
Schmid RD, Verger R (1998) Esterases: interfacial enzymes with attractive applications. Angew Chem Int Ed 37:1608–1633
Schmidt-Dannert C, Schmid R (1996) Thermoalkalophilic esterase of Bacillus thermocatenulatus. 1. molecular cloning, nucleotide sequence, purification and some properties. Biochim Biophys Acta 1301:105–104
Schmidt-Dannert C, Rua ML, Wahl S, Schmid RD (1997) Bacillus thermocatenulatus esterase: a thermoalkalophilic esterase with interesting properties. Biochem Soc Trans 25:178–182
Sharma R, Chisti I, Chand BU (2001) Production, purification, characterization and applications of esterases. Biotechnol Adv 19:627–662
Siezen R, de Vos WM, Leunissen JA, Dijkstra BW (1991) Homology modelling and protein engineering strategy of subtilases, the family of subtilisin-like serine proteinases. Protein Eng. 4(7):719–737
Sinchaikul S, Sookkheo B, Phutrakul S, Pan F, Chen S (2001) Optimization of a thermostable esterase from Bacillus stearothermophilus P1: overexpression, purification and characterization. Protein Expr Purif 22:388–398
Sroga GE, Dordick JS (2001) Generation of a broad esterolytic subtilisin using combined molecular evolution and periplasmic expression. Protein Eng 14(11):929–937
Stocklein W, Sztajer H, Menge U, Schmid R (1993) Purification and properties of an esterase from Penicillium expansum. Biochim Biophys Acta 1168:181–189
Sugihara A, Shimada Y (1990) Separation and characterization of two molecular forms of Geotrichum candidum esterase. J Biochem 100:1207–1213
Sugihara A, Ueshima M, Shimada Y, Tsunasawa S, Tominaga Y (1992) Purification and characterization of a novel thermostable esterase from Pseudomonas cepacia. J Biochem 112:598–603
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76(9):4350–4354
Yabuta Y, Takagi H, Inouye M, Shinde U (2001) Folding pathway mediated by an intramolecular chaperone: propeptide release modulates activation precision of pro-subtilisin. J Biol Chem 276:44427–44434
Yabuta Y, Subbian E, Oiry C, Shinde U (2003) Folding pathway mediated by an intramolecular chaperone. A functional peptide chaperone designed using sequence databases. J Biol Chem 78:15246–15251
Acknowledgments
This work was partially supported by IFS. CPM received a scholarship from CONACyT, Mexico. We thank Dr. Guillermo Mendoza for the N-terminal amino acid sequencing of the enzyme. We thank Norma Ballesteros for the activity assays on protease substrates. We thank M. in C. Idalia Flores for technical assistance. We appreciate the English revision by American Journal Experts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peña-Montes, C., González, A., Castro-Ochoa, D. et al. Purification and biochemical characterization of a broad substrate specificity thermostable alkaline protease from Aspergillus nidulans . Appl Microbiol Biotechnol 78, 603–612 (2008). https://doi.org/10.1007/s00253-007-1324-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1324-y