
Abstract
Debaryomyces castellii phytase was purified to homogeneity in a single step by hydrophobic interaction chromatography. Its molecular mass is 74 kDa with 28.8% glycosylation. Its activity was optimal at 60°C and pH 4.0. The K m value for sodium phytate was 0.532 mM. The enzyme exhibited a low specificity and hydrolyzed many phosphate esters. The phytase fully hydrolyzed myo-inositol hexakisphosphate (or phytic acid, Ins P6) to inositol and inorganic phosphate. The sequence of Ins P6 hydrolysis was determined by combining results from high-performance ionic chromatography and nuclear magnetic resonance. D. castellii phytase is a 3-phytase that sequentially releases phosphate groups through Ins (1,2,4,5,6) P5, Ins (1,2,5,6) P4, Ins (1,2,6) P3, Ins (1,2) P2, Ins (1 or 2) P1, and inositol (notation 3/4/5/6/1 or 2).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phytic acid (myo-inositol hexakisphosphate) is the major storage form of phosphorus in cereals and leguminous plants (Reddy et al. 1982). Two features of phytic acid are of particular interest in human and animal food (Wodzinski and Ullah 1996): (1) phytic acid is only slightly broken down within the digestive tract of monogastric animals hence requiring food products to be complemented with inorganic phosphate, and (2) phytic acid is considered as an antinutritional and chelating agent that reduces the availability of proteins and ions (Fe3+, Ca2+, Zn2+, Mg2+).
Phytases (myo-inositol hexakisphosphate 3- and 6-phosphohydrolases; EC 3.1.3.8 and EC 3.1.3.26) catalyze the hydrolytic degradation of myo-inositol hexakisphosphate (phytic acid, Ins P6) to free inorganic phosphate (Pi), to yield lower myo-inositol phosphate esters (Ins P5 to Ins P1) and, in some cases, free myo-inositol.
Phytases are produced by a wide range of organisms: plants, animals, and particularly fungi, bacteria, and yeasts (Shieh and Ware 1968; Nayini and Markakis 1984; Nakamura et al. 2000; Pandey et al. 2001; Vohra and Satyanarayana 2003).
The increasing use of phytases as additives in many biotechnological applications, including animal feed, reinforces the interest of: (1) isolating new and efficient phytase-producing microorganisms, (2) obtaining effective phytases capable of releasing as many of the food phosphates as possible into the digestive tract, and (3) selecting phytases that remain stable during food processing and storage, with the lowest production costs.
Investigations carried out in our laboratory since 1990 have allowed us to select phytase-producing yeasts (Lambrechts et al. 1992). In the present work, we describe the production of a new phytase from the previously selected yeast Debaryomyces castellii CBS 2923.
Following purification, biochemical features and specificity of hydrolysis of Ins P6 by D. castellii phytase are described. The stereospecificity of hydrolysis of Ins P6 by D. castellii phytase was established. The structure of intermediate myo-inositol phosphates was analyzed by chromatography and 1D- and 2D-nuclear magnetic resonance (NMR), and the final product was identified.
Materials and methods
Strain and growth conditions
The strain used is kept under the name D. castellii CBS 2923 at the CentraalBureau voor Schimmelculture (Delft, Holland). Cultures were performed in low inorganic phosphate content synthetic medium (MSA) (Lambrechts et al. 1992).
Purification
The culture supernatant obtained after centrifugation (7,150 × g, 15 min, 4°C; Beckman J21B, JA14 rotor) was filtered on a 0.22-μm cutoff membrane and concentrated by tangential flow ultrafiltration on a 10-kDa cutoff membrane in an Ultrassette™ device (Pall Corporation, Northborough, MA). The concentrated enzyme was applied to a HiPrep Phenyl FF column (Amersham Pharmacia Biotech). Proteins were eluted with a linear (NH4)2SO4 gradient from 2 to 0 M. Phytase was eluted with 1.7 M (NH4)2SO4. The flow rate was 5 mL/min. The absorbance was measured at 280 nm. Fractions containing phytase were pooled, desalted by ultrafiltration through a PM 10 poly-l-lactide-co-glycolide-co-ɛ-caprolactone membrane, and concentrated. Protein purity was checked using sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE). Protein concentration was measured by Bradford’s method (Bradford 1976) with bovine serum albumin as standard.
Electrophoresis
SDS-PAGE and native-PAGE electrophoreses were performed on precast gradient 4–15% Tris–HCl polyacrylamide gels (Biorad). Proteins were visualized by Coomassie blue staining and quantified by ImageMaster TotalLab software (Amersham Biosciences). Samples (100 μg) were deglycosylated during 2 h at 37°C using 250 U of endo-β-N-acetylglycosaminidase H (Endo H, New England Biolabs, Beverly, MA). Specific phytase revelation was performed by incubating gels in 100 mL revelation solution containing 2 g/L α-naphtylphosphate, 1 g/L FastGarnet GBC, 0.92 g/L sodium phytate, buffered at pH 5.5 using 0.25 M sodium acetate buffer.
Determination of molecular mass by mass spectrometry
Analysis was carried out on the SDS-PAGE-purified phytase with a spectrometer Matrix-assisted laser desorption/ionization time of flight Biflex III scout 384 (Bruker, Breme, Germany). Analyses were performed by the Protéome platform (Languedoc-Roussillon Génopole, ENSAM-INRA, Montpellier).
Phytase activity and properties
Phytase activity was determined by following the liberation of inorganic phosphate using the ferrous sulfate–molybdenum blue method (Ullah and Gibson 1987). Activity was measured in the presence of 8 mM sodium phytate (in 200 mM sodium acetate buffer, 1 mM CaCl2, pH 4.0) at 37°C and using a Beckman DU530 spectrophotometer (700 nm). One phytase activity unit (U) was defined as the amount of enzyme that releases 1 μmol of inorganic phosphate from sodium phytate per min under the defined reaction conditions.
Optimal pH of activity was determined (37°C; n = 3) by using buffers (200 mM) of glycine–HCl, (pH 2.0, 2.5, 3.0, 3.5), sodium acetate–acetic acid (pH 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0), and Tris–HCl (pH 7.0, 7.5). Kinetics were performed at different temperatures (30 to 80°C) to determine the optimal temperature at pH 4.0 (n = 3). Thermostability was determined (n = 3) by incubating the enzyme sample in water or in 125 mM sodium acetate buffer, 1 mM CaCl2, pH 4.0, at 60, 65, 66.5, 70, and 75°C during 5, 10, 15, 20, 40, 60, and 120 min. After heat treatment, samples were chilled on ice and the remaining phytase activity was measured at 37°C as described above. pH and temperature stability was estimated (n = 3) by incubating the enzyme sample for 1 h at 40 or 60°C or 67 days at −20°C in the same buffer solutions as those used in the study of the effect of pH. After treatment, samples were chilled on ice, and the remaining phytase activity was measured at 37°C as described above. K m was determined at 37°C by incubation of the enzyme (100 mU) with sodium phytate (pH 4.0) or p-nitrophenylphosphate (p-NPP; pH 5.5; 0.1 to 6 Mm) in sodium acetate buffer (250 mM). Initial velocities were calculated from linear regions of the hydrolysis curve and plotted against the inorganic phosphate concentration. Linear transformation was achieved using Lineweaver–Burk plots to estimate K m parameter. The effect of cations on phytase activity was determined under standard assay conditions.
Stereospecificity studies
Hydrolysis reactions were performed at 40°C, pH 4.0. Enzyme (3 U/mL, 1 vol) was added to the medium (4 vol, 8 mM sodium phytate in 250 mM sodium acetate buffer) to start the reaction. Reactions were stopped after different incubation times up to 6 h by denaturing the enzyme at 100°C during 10 min. We checked that the different inositol phosphates remained unchanged. Before injection, samples (1 vol) were mixed with 0.05 M HCl (1 vol). For identification of peaks, a reference sample was prepared by chemical hydrolysis by dissolving 50 mg of sodium phytate in 5 mL of 6 M HCl. The solution was incubated 16 h at 100°C and evaporated to dryness. Then, 20 mL of 25 mM HCl were added to the hydrolyzed sample.
Inositol phosphate content was determined using high-performance ionic chromatography (HPIC). The chromatographic system and method were according to the first system developed by Skoglund et al. (1997, 1998). The chromatograph was equipped with a 100-μL injector loop. This method allows the separation of Ins P6 to Ins P1 and the identification of 12 isomers of Ins P5 and Ins P4. The elution order of peaks after chemical hydrolysis of phytate was established by Türk et al. (2000).
NMR characterization of inositol phosphates
Ins P5, Ins P4, Ins P3, Ins P2, and Ins P1 were isolated by HPIC, pooled, and lyophilized. Samples (50–200 μg) were then solubilized in 500 μL of deuterated water (D2O) for NMR analysis.
1H-NMR experiments were performed on Bruker Avance 600 and Avance 500 spectrometers equipped with a triple-resonance cryoprobe (1H, 13C, and 15N) and z-gradients. The 1H-NMR spectra with 31P decoupling and 1H–31P heteronuclear multiple quantum correlation (HMQC) spectra were recorded on a Bruker Avance 400 equipped with a triple broadband inverse probe. The residual water resonance was suppressed by using a low-power irradiation. 1H chemical shifts were referenced with respect to sodium (trimethylsilyl)-3-propionate-d4 (TSP, 0 ppm) or to the residual signal of water set to 4.914 ppm at 17°C. For the assignment of proton resonance, the double-quantum filtered-correlated spectroscopy (DQF-COSY; Derome and Williamson 1990; Rance et al. 1983) and z-filtered total-correlated spectroscopy (TOCSY; Bax and Davis 1985; Rance 1987) were recorded by using 512 t1 increments. A 50-ms spinlock was used for the TOCSY. 1H–31P HMQC spectra were recorded with 64 t1 increments (Bax et al. 1983). Data were processed by using the XWINNMR software (Bruker BioSpin, Wissembourg, France).
Proton assignments were obtained from analysis of the DQF-COSY and TOCSY data. When the proton resonance was obtained, phosphorylated positions were unambiguously determined both from spectra obtained with the 31P decoupling or 1H–31P HMQC spectra.
1H-NMR monitoring of hydrolysis kinetics
To follow the hydrolysis kinetics, 3.8 mM Ins P6 samples in phosphate buffer (28 mM) were prepared in D2O. TSP (0 ppm) was used as the internal reference. Fifteen milliunits of phytase were added to the Ins P6 sample. From the typical spectra of each inositol phosphate previously obtained, their formation and disappearance as well as the formation of inositol were monitored during hydrolysis.
Results
Physical characteristics
The phytase was purified to apparent homogeneity (SDS-PAGE, Fig. 1) in a single step by hydrophobic interaction chromatography. Purification factor and yield were 13.6 and 59%, respectively. Specific activity of the purified extract was 157.4 U/mg.

SDS-PAGE (a) and zymogram (b) analysis of phytase of D. castellii before (lanes 2, 4, 6) and after (lanes 1, 3, 5) deglycosylation by Endo H. Lanes 5, 6: specific revelation. Lane M: standard protein (Pharmacia Biotech)
The molecular mass of the phytase was estimated, by SDS-PAGE electrophoresis or mass spectrometry, to 77 or 74.46 kDa, respectively, for the glycosylated enzyme and 51 or 52.98 kDa, respectively, after deglycosylation. Under nondenaturing conditions, apparent molecular mass was estimated to 200–400 kDa for the glycosylated form. Specific revelation showed that the deglycosylated phytase was active (Fig. 1).
Biochemical characteristics
The phytase was active for pH values between 2.5 and 6.5, with an optimal pH between 4.0 and 4.5. The nature of the buffer had an impact on phytase activity; at pH 3.5, the sodium acetate buffer had an inhibitory effect (30%) in comparison to the glycine buffer.
The phytase activity was optimal between 55 and 60°C. The activation energy was 38,000 J/mol. The study of thermal denaturation showed that the enzyme was stable for 1 h at 60°C in water or 1 h at 66°C in sodium acetate buffer (125 M, pH 4.0). The phytase was denatured when temperature exceeded 68°C, with a 70% activity loss after 1 hour at 70°C. The activation energy for denaturation was 606,000 J/mol.
The combined effect of temperature and pH on enzyme denaturation over time was also assayed. No denaturation was observed for pH values between 5.0 and 7.0 after 67 days of storage at −20°C, whereas the phytase was denatured for pH values lower than 5.0. For pH values ranging from 3.0 to 7.0, the enzyme retained 80 to 100% of its activity after incubation at 40°C for 1 h.
Among various cations tested (5 mM), Ca2+ and Fe3+ induced a 10% reduction in enzyme activity. In the presence of Co2+, Zn2+, Cu2+, and Mg2+, the activity was inhibited by 31 to 58%, and Mn2+ induced a strong inhibition (71%). Although calcium had no direct influence on enzymatic activity, it played a crucial protective role. In the absence of calcium, the denaturation of the enzyme was complete after 1 h at 66.5°C.
Kinetic constants were determined for two substrates: sodium phytate and p-NPP. The enzyme’s affinity was four times stronger for sodium phytate (K m = 0.532 mM, K cat = 51.2 s−1) than for p-NPP (K m = 2.27 mM, K cat = 44.2 s−1). The enzyme was inhibited by phosphates with a K i of 1.3 mM. This phytase was characterized by a wide activity spectrum, with preferential hydrolysis of sodium phytate (100%, 9 mM), p-NPP (135%, 4 mM), phosphoenolpyruvate (130%), adenosine triphosphate (133%), and adenosine diphosphate (120%). It can also hydrolyze various phosphorylated substrates such as adenosine monophosphate (50%), glucose-6-phosphate (82%), fructose-6-phosphate (35%), α-glycerophosphate (78%), 3-phosphoglyceric acid (95%), or several inositol phosphate, in particular Ins(2)P1 (84%, 1 mM) and Ins(1)P1 (58%).
Stereospecificity of myo-inositol hexakisphosphate dephosphorylation by the phytase
The isolation of the different Ins Pn intermediates (Ins P6 to Ins P1) and the identification of Ins P5 and Ins P4 isomers were performed using HPIC. The various isomers were identified by comparing chromatograms obtained from the hydrolysis by D. castellii phytase with those: (1) described in the literature and obtained under similar experimental conditions (Skoglund et al. 1997), (2) obtained by chemical hydrolysis under the conditions described by Türk et al. (2000), and (3) obtained from the hydrolysis of phytic acid by Aspergillus niger and Peniophora lycii phytases (Lassen et al. 2000). The formation and disappearance over time of inositol phosphates produced during the hydrolysis of phytic acid by the D. castellii phytase were monitored (Fig. 2). Fifteen minutes after the beginning of hydrolysis, Ins P6 had almost completely disappeared, and three major peaks corresponding to Ins(1,2,4,5,6)P5, Ins(1,2,5,6)P4, and several Ins P3 peaks were detected. The attribution of Ins P5 and Ins P4 was confirmed by the addition of authentic inositol samples (data not shown). After 300 min, the amount of free phosphates measured corresponded to 100% of the potential phosphate available from the phytate molecule (data not shown). This phytase hydrolyzes and releases all the phosphate groups. Characterization of the Ins Pn fractions, separated by chromatography, was completed by NMR.

HPIC analysis of inositol phosphates formed from chemical phytate hydrolysis (a) and Ins P6 hydrolysis for 15 (b), 75 (c), and 120 min (d) by phytase of D. castellii. 1 Ins P1 and Pi, 2 Ins P2, 3 Ins P3, 4 Ins (1,2,5,6) P4, 5 Ins (1,2,4,5,6) P5, 6 Ins P6
Kinetics of Ins P6 monitored by 1H NMR
As described in “Materials and methods,” the purified fractions of Ins P1, Ins P2, Ins P3, and Ins P4 were characterized by their 1H- and 31P–1H (HMQC) spectra. Among the 63 theoretically possible diastereoisomers, the 24 enantiomer pairs cannot be distinguished by NMR. Thus, the identification of the hydrolysis products did not allow us to distinguish a 1-phytase from a 3-phytase and a 4-phytase from a 6-phytase. Conventionally, when positions 1 or 3 are first hydrolyzed, the phytase is referred to as a 3-phytase. Similarly, when positions 4 or 6 are first hydrolyzed, the phytase is referred to as a 6-phytase.
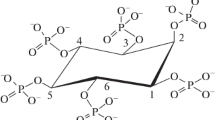
Preliminary studies revealed that the phytase activity was insensitive to deuterated water. The kinetics of Ins P6 hydrolysis shows the successive formation of Ins P5, Ins P4, Ins P3, Ins P2, Ins P1, and inositol (Fig. 3). From these data, we can deduced the entire phosphate groups hydrolysis sequence (Fig. 4): Ins(1,2,4,5,6)P5 is the first intermediate produced and is rapidly hydrolyzed into Ins(1,2,5,6)P4 and then into Ins(1,2,6)P3. This latter accumulates and is further hydrolyzed into Ins(1,2)P2. These results show that phytase has a weaker affinity for Ins P3 than for Ins P6, Ins P5, and Ins P4 and confirmed the results obtained by HPIC. The last two ester bonds are hydrolyzed simultaneously, which results in the formation of inositol. These results confirmed the ability to hydrolyze inositol monophosphates such as Ins (2) P1 and Ins (1) P1. These data clearly show all six phosphate bonds have been hydrolyzed.

Set of spectra recorded at 600 MHz and 17°C for a kinetic yielding the inositol as the final product (1.7 mM Ins P6, 10 mM acetate buffer, pH 4.0, H2O/D2O, 16/84 v/v, 15 mU of phytase). 400 spectra recorded for 20 h, 30 min between two displayed spectra. Only some typical signals are labeled

Kinetic of Ins P6 hydrolysis by D. castellii phytase monitored by NMR (1.7 mM Ins P6, 10 mM acetate buffer, pH 4.0, H2O/D2O, 16/84 v/v, 15 mU of phytase, 600 MHz)
Discussion
To our knowledge, the D. castellii phytase has never been described so far. Both glycosylated and deglycosylated forms are active. Most phytases of bacterial or fungal origin are monomers (Wyss et al. 1999a). Some phytases, however, are described as oligomers: the Schwanniomyces castellii CBS 2863 (syn. Debaryomyces occidentalis) phytase is a pentamer (Segueilha et al. 1992), that of Aspergillus terreus is an hexamer (Yamamoto et al. 1972), while A. niger pH 2.5 acid phosphatase (AnigAP) is a tetramer (Kostrewa et al. 1999). A structural study has confirmed that D. castellii phytase is a tetramer (unpublished data).
Biochemical features are similar to fungal or yeast phytases (Table 1). Only a few natural phytases remain stable at temperatures above 60°C. Such phytases include the Bacillus sp. DS11 phytase, which retains 100% residual activity after 10-min incubation at 70°C (Kim et al. 1998) or the S. castellii phytase that remains stable for 60 min at 70°C (Segueilha et al. 1992). The D. castellii phytase remained stable for temperatures up to 60°C for pH values ranging from 3.0 to 7.0. Its activity and stability, however, were reduced in the presence of acetate ions.
The affinity of the D. castellii phytase toward phytate (K m = 0.532 mM) is similar to the Bacillus subtilis, Escherichia coli, Klebsiella terrigena, and wheat phytases (Table 1). In contrast, the affinity of Aspergillus ficuum, Aspergillus fumigatus, P. lycii, and S. castellii phytases is approximately ten times stronger (0.040 mM). On the contrary to phytases from A. niger (Phyt B), Klebsiella aerogenes, Pseudomonas, Bacillus, and E. coli with a narrow spectrum, the D. castellii phytase has a wide spectrum of activity and thus belongs to the same group as A. ficuum, wheat (spelt variety or Triticum spelta), A. fumigatus, S. castellii phytases, or AnigAP (Wyss et al. 1999b).
Chromatographic analysis as well as homo- or heteronuclear NMR analyses allow us to determine without ambiguity the structure of the different intermediates produced during phytic acid hydrolysis: Ins P5, Ins P4, Ins P3, Ins P2, and Ins P1. HPIC analysis allowed to establish that phosphate groups in positions 3 then 4 were the first hydrolyzed and thus this phytase belongs to the 3-phytase family (EC 3.1.3.8), which includes phytases from many other microorganisms. Kinetics monitored by NMR allowed us to determine the release sequence of phosphate groups through Ins (1,2,4,5,6) P5, Ins (1,2,5,6) P4, Ins (1,2,6) P3, Ins (1,2) P2, Ins (1 or 2) P1, and inositol (notation 3/4/5/6/1 or 2). The two methods used (HPIC or NMR) show that the phytase of the yeast D. castellii is able to hydrolyze the six phosphate bonds. Among the known phytases, very few have the capacity to fully hydrolyze phytic acid. Fungal phytases from A. niger, A. fumigatus, Emericella nidulans, A. terreus, P. lycii, or from Saccharomyces cerevisiae can only hydrolyze phosphate groups in equatorial position. The phosphate group in position 2, which is in an axial position, is never hydrolyzed (Lassen et al. 2000; Wyss et al. 1999b; Greiner et al. 2001). E. coli phytase releases only five phosphates, but in some rare instances, traces of inositol or Ins (1) P1 can be detected (Greiner et al. 2000). In Bacillus amyloliquefaciens, the final products were restricted to Ins P3 (Kerovuo et al. 2000).
The properties of the D. castellii phytase (stability, thermostability, broad pH range for activity, ability to fully hydrolyze the phytic acid) make it an attractive enzyme for use in the animal feed industry. To our knowledge, it is the first yeast phytase described so far as capable of achieving full phytic acid hydrolysis and for which the full sequence of hydrolysis has been characterized. To explain this originality, work is continuing on complex crystals of this phytase with the end product.
References
Bax A, Griffey RH, Hawkins BL (1983) Correlation of proton and nitrogen-15 chemical shifts by multiple quantum RMN. J Magn Reson 55:301
Bax A, Davis GD (1985) MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J Magn Reson 65:355–360
Bradford M (1976) A rapid and sensitive method for the quantification of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Derome AE, Williamson MP (1990) Rapid-pulsing artefacts in double-quantum-filtered COSY. J Magn Reson 88:177–185
Greiner R, Konietzny U, Jany KD (1993) Purification and characterization of two phytases from E. coli. Arch Biochem Biophys 303:107–113
Greiner R, Haller E, Konietzny U, Jany KD (1997) Purification and characterization of a phytase from Klebsiella terrigena. Arch Biochem Biophys 341:201–206
Greiner R, Carlsson NG, Alminger ML (2000) Stereospecificity of myo-inositol hexakisphosphate dephosphorylation by a phytate-degrading enzyme of Escherichia coli. J Biotechnol 84:53–62
Greiner R, Alminger ML, Carlsson NG (2001) Stereospecificity of myo-inositol hexakisphosphate dephosphorylation by a phytate-degrading enzyme of Baker’s yeast. J Agric Food Chem 49:2228–2233
Kerovuo J, Rouvinen J, Hatzack F (2000) Analysis of myo-inositol hexakisphosphate hydrolysis by Bacillus phytase: indication of a novel reaction mechanism. Biochemistry 352:623–628
Kim YO, Kim HK, Bae KS, Yu JH, Oh TK (1998) Purification and properties of a thermostable phytase from Bacillus sp. DS11. Enzyme Microb Technol 22:2–7
Kostrewa D, Wyss M, D’Arcy A, van Loon APGM (1999) Crystal structure of Aspergillus niger pH 2.5 acid phosphatase at 2.4 Å resolution. J Mol Biol 288:965–974
Lambrechts C, Boze H, Moulin G, Galzy P (1992) Utilization of phytase by some yeasts. Biotechnol Lett 14:61–66
Lassen SF, Bech L, Fuglsang CC, Ohmann A, Breinholt J, Østergaard PR (2000) Peniophora phytase. US Patent 6060298
Lassen SF, Breinholt J, Østergaard PR, Brugger R, Bischoff A, Wyss M, Fuglsang CC (2001) Expression, gene cloning and characterization of five novel phytases from four basidiomycete fungi: Peniophora lycii, Agrocybe pediades, a Ceriporia sp., and Trametes pubescens. Appl Environ Microbiol 67:4701–4707
Nagashima T, Tange T, Anazawa H (1999) Dephosphorylation of phytase by using the Aspergillus niger phytase with a high affinity for phytate. Appl Environ Microbiol 65:4682–4684
Nakamura Y, Fukuhara H, Sano K (2000) Secreted phytase activities of yeast. Biosci Biotechnol Biochem 64:841–844
Nayini NR, Markakis P (1984) The phytase of yeast. Lebensm Wiss Technol 17:24–26
Pandey A, Szakács G, Soccol CR, Rodriguez-Leon JA, Soccol VY (2001) Production, purification and properties of microbial phytases. Bioresour Technol 77:203–214
Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wüthrich K (1983) Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Commun 117:479–485
Rance M (1987) Improved techniques for homonuclear rotating-frame and isotropic mixing experiments. J Magn Reson 74:557–564
Reddy NR, Sathe SK, Salunkhe DK (1982) Phytases in legumes and cereals. Adv Food Res 28:1–92
Segueilha L, Lambrechts C, Boze H, Moulin G, Galzy P (1992) Purification and properties of the phytase from Schwanniomyces castellii. J Ferm Bioeng 74:7–11
Shieh TR, Ware JH (1968) Survey of microorganisms for the production of extracellular phytase. Appl Microbiol 16:1348–1351
Skoglund E, Carlsson NG, Sandberg AS (1997) Determination of isomers of inositol mono- to hexaphophates in selected foods and intestinal contents using high-performance ion chromatography. J Agric Food Chem 45:431–436
Skoglund E, Carlsson NG, Sandberg AS (1998) High-performance chromatographic separation of inositol phosphate isomers on strong anion exchange columns. J Agric Food Chem 46:1877–1882
Türk M, Sandberg AS, Carlsson NG, Andlid T (2000) Inositol hexaphosphate hydrolysis by baker’s yeast. Capacity, kinetics, and degradation products. J Agric Food Chem 48:100–104
Ullah AHJ, Gibson DM (1987) Extracellular phytase (E.C. 3.1.3.8.) from Aspergillus ficuum NRRL 3135: purification and characterization. Prep Biochem 17:63–91
Ullah AHJ, Sethumadhavan K, Lei XG, Mullaney EJ (2000) Biochemical characterization of cloned Aspergillus fumigatus phytase. Biochem Biophys Res Commun 275:279–285
Ullah AHJ, Sethumadhavan K (2003) PhyA gene product of Aspergillus ficuum and Peniophora lycii produces dissimilar phytases. Biochem Biophys Res Commun 303:463–468
Vohra A, Satyanarayana T (2003) Phytases: microbial sources, production, purification and potential biotechnological applications. Crit Rev Biotechnol 23:29–60
Wodzinski RJ, Ullah AHJ (1996) Phytase. Adv Appl Microbiol 42:263–302
Wyss M, Pasamontes L, Friedlein A, Rémy R, Tessier M, Kronenberger A, Middendorf A, Lehmann M, Schnoebelen L, Röthlisberger U, Kusznir E, Wahl G, Müller F, Lahm HW, Vogel K, van Loon APGM (1999a) Biophysical characterisation of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): molecular size, glycosylation pattern, and engineering of proteolytic resistance. Appl Environ Microbiol 65:359–366
Wyss M, Brugger R, Kronenberger A, Rémy R, Fimbel R, Oesterhelt G, Lehmann M, van Loon APGM (1999b) Biochemical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolase): catalytic properties. Appl Environ Microbiol 65:367–373
Yamamoto M, Minoda Y, Yamada K (1972) Chemical and physico-chemical properties of phytase from Aspergillus terreus. Agric Biol Chem 36:2097–2103
Acknowledgments
This work was supported by ADISSEO France SAS society.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ragon, M., Aumelas, A., Chemardin, P. et al. Complete hydrolysis of myo-inositol hexakisphosphate by a novel phytase from Debaryomyces castellii CBS 2923. Appl Microbiol Biotechnol 78, 47–53 (2008). https://doi.org/10.1007/s00253-007-1275-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1275-3