
Abstract
Because engineering of the 101.016-bp megaplasmid pKB1 of Gordonia westfalica Kb1 failed due to the absence of an effective transfer system, pKB1 was transferred by conjugation from G. westfalica Kb1 to a kanamycin-resistant mutant of Rhodococcus opacus PD630 at a frequency of about 6.2 × 10−8 events per recipient cell. Furthermore, pKB1 was transferred to G. polyisoprenivorans strains VH2 and Y2K and to Mycobacterium smegmatis by electroporation at frequencies of 5.5 × 103, 1.9 × 103, and 8.3 × 102 transformants per microgram plasmid DNA. The pKB1-encoded cadmium resistance gene cadA was used for selection in these experiments. Recombinant pKB1-containing G. polyisoprenivorans VH2 and M. smegmatis were then used to engineer pKB1. A kanamycin resistance cassette was inserted into the pKB1-encoded cadA gene, ligated to suicide plasmid pBBR1MCS-5, and the resulting plasmid was electroporated into plasmid-harboring strains. Homologous recombination between cadA on suicide plasmid and the respective sequence in pKB1 led to its integration into pKB1. Thus, two selection markers were accommodated in pKB1 to monitor plasmid transfer into Gordonia and related taxa for analysis of genes essential for rubber degradation and others. In this study, two transfer methods for large plasmids and strategies for engineering of pKB1 were successfully applied, thereby, extending the tool box for Gordonia.
Similar content being viewed by others

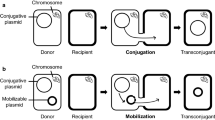
Avoid common mistakes on your manuscript.
Introduction
Gordonia sp. are Gram-positive bacteria belonging to the so-called CMN group (Corynebacterium, Mycobacterium, and Nocardia) of actinomycetes that possess a variety of interesting metabolic capabilities (Arenskötter et al. 2004). Several members of the genus Gordonia exhibit special degradation activities such as degradation of poly(cis-1,4-isoprene) by G. westfalica strain Kb1 (Linos and Steinbüchel 1998; Linos et al. 1999, 2002) and G. polyisoprenivorans (Linos and Steinbüchel 1998; Arenskötter et al. 2001); desulfurization of benzothiophene by G. desulfuricans (Gilbert et al. 1998; Kim et al. 1999) or dibenzothiophene by G. amicalis (Kim et al. 2000); biodegradation of ethyl t-butyl ether (ETBE), methyl t-butyl ether (MTBE), and t-amyl methyl ether (TAME) by G. terrae (Hernandez-Perez et al. 2001); degradation of 3-ethylpyridine and 3-methylpyridine by G. nitida (Yoon et al. 2000); and degradation of propane by Gordonia sp. strain TY-5 (Kotani et al. 2003, 2007).
There is still a lack of suitable genetic tools to apply genetic engineering and recombinant DNA techniques to members of this interesting group of bacteria. Recently, Escherichia coli/Rhodococcus shuttle vectors suitable for transfer of foreign DNA to members of the genus Gordonia were described (Arenskötter et al. 2003). The origin of replication of a circular 101,016-bp megaplasmid pKB1 from the poly(cis-1,4-isoprene)-degrading bacterium G. westfalica strain Kb1 was identified and used for construction of E. coli/Gordonia shuttle vectors suitable for gene cloning and expression in several Gordonia species and members of related taxa (Bröker et al. 2004). One of these constructed E. coli/Gordonia shuttle vectors and an E. coli/Rhodococcus shuttle vector were suitable for transformation of G. jacobae by electroporation (Veiga-Crespo et al. 2006). Little is also known about vectors appropriate for transposon mutagenesis or for generation of knock-out mutants in Gordonia sp. Stable Tn5096-based transposon mutagenesis in G. polyisoprenivorans strain VH2 was only recently established (Banh et al. 2005). Although the thermosensitive plasmid pCG79 could be transferred to G. polyisoprenivorans strain VH2, indicating stable propagation of the plasmid, Tn611-based transposon mutagensis in the same strain yielded unstable mutants reverting to a wild-type phenotype (Banh et al. 2005). Others described the insertion of a plasmid by homologous recombination between a cloned sequence within the plasmid and the corresponding sequence in the chromosomes of G. amarae (Dogan et al. 2006) and G. polyisoprenivorans strain VH2 (Arenskötter et al., unpublished data).
Because evidence was obtained that genes essential for rubber degradation are encoded by pKB1 (Bröker et al. 2004), the need for manipulation of this plasmid arose. Several attempts to engineer pKB1 in G. westfalica strain Kb1 failed because of the absence of an effective gene transfer system for this strain. To engineer plasmid pKB1, it has to be transferred to strains that are genetically approachable. To monitor successful transfer of pKB1 into related strains, a pKB1-specific selection marker had to be identified. Attention was drawn to the pKB1-encoded gene cadA (ORF34). Its translational product was related to a putative Cd2+/Zn2+-transporting P-type ATPase, and its heterologous expression conferred cadmium but not zinc resistance to E. coli strain RW3110 (Bröker et al. 2004). In this study, it was investigated whether cadA is a useful selection marker for transfer of pKB1 into cadmium-sensitive strains of Gordonia and related taxa by electroporation and conjugation. The various translational products of a large region comprising putative conjugative transfer genes located on pKB1 should enable conjugative transfer of pKB1 from the natural host G. westfalica strain Kb1 as donor to related bacteria in which this megaplasmid is replicated. For example, ORF67 presumably encodes a TraA-like protein with 31% identity to TraA encoded by megaplasmid pREA400 of R. erythropolis strain AN12; the latter was recently demonstrated to be essential for conjugation (Yang et al. 2007). DNA relaxases encoded by traA genes are the main components for initiation of conjugative plasmid transfer because they catalyze the cleavage of one plasmid strand at the nic site within the oriT via a transesterification reaction. Transfer of the nicked plasmid to the recipient cell occurs unidirectional in the form of a single-stranded DNA intermediate (for reviews, see references Pansegrau and Lanka 1996, Byrd and Matson 1997, and Grohmann et al. 2003).
In this study, we demonstrated the transfer of megaplasmid pKB1 to genetically approachable cadmium-sensitive strains of Gordonia and related taxa by conjugation and also by electroporation using cadA as selection marker. Furthermore, genetic manipulation of pKB1 in the resulting recombinant strains of G. polyisoprenivorans strain VH2 and M. smegmatis strain mc2155 by integration of a suicide vector into plasmid pKB1 was successfully demonstrated. This yielded a recombinant plasmid pKB1 with two useful additional antibiotic selection markers (kanamycin and gentamycin resistance) to monitor transfer of this plasmid into other cadmium-resistant strains for determination of new plasmid-encoded features.
Materials and methods
Bacterial strains, plasmids, and cultivation conditions
Bacteria and plasmids used in this study are listed in Table 1. All strains of the genus Gordonia and Rhodococcus opacus strain PD630 were grown at 30°C on standard I complex nutrient broth (St-I, Merck, Darmstadt, Germany) or mineral salts medium (MSM; Schlegel et al. 1961). Mycobacterium smegmatis strain mc2155 was grown at 30°C on Luria Bertani (LB) broth (Sambrook et al. 1989) containing 0.05% (w/v) Tween 80 or mineral salts medium (MSM; Schlegel et al. 1961). Carbon sources were added to liquid MSM as indicated in the text. Cells of E. coli strains were cultivated at 37°C in LB broth. Antibiotics were applied according to Sambrook et al. (1989) and as indicated in the text. Liquid cultures were done in Erlenmeyer flasks and incubated on a horizontal rotary shaker at 180 rpm. Solid media were prepared by addition of agar-agar (15 g/l).
Kanamycin-resistant mutant of R. opacus strain PD630
For selection of a spontaneous kanamycin resistant mutant of R. opacus strain PD630, 50 ml St-I medium in 300-ml Erlenmeyer flasks were inoculated with cells of this strain and incubated at 30°C. After 2 days incubation, 50 μl cell suspensions were used to inoculate Erlenmeyer flasks with fresh St-I medium containing a slightly increased kanamycin concentration. After successive increase of the kanamycin concentration from 0 to 100 μg/ml, cells deriving from a culture containing the highest kanamycin concentration were diluted and spread on St-I agar plates containing 100 μg/ml kanamycin. This procedure yielded the spontaneous kanamycin-resistant mutant PD630Km100, which was used as recipient for conjugative transfer of megaplasmid pKB1.
Determination of metal tolerance
For the recombinant, pKB1-harboring Gram-positive bacteria of the genera Gordonia, Rhodococcus, and Mycobacterium, St-I agar plates containing 100 to 800 μM CdCl2 were prepared using a 0.1-M stock solution of CdCl2 · H2O (Merck), which was sterilized by filtration through a 0.22-μm membrane filter (Roth, Karlsruhe, Germany). The plates were inoculated, and tolerance for cadmium was evaluated after 3 days incubation at 30°C.
Isolation, analysis, and modification of DNA
Plasmid DNA was prepared from crude lysates by the alkaline extraction method (Birnboim and Doly 1979). Before lysis, cells of Gordonia, Rhodococcus, and Mycobacterium were incubated in the presence of lysozyme (2 mg/ml) for 2 h at 37°C. DNA was restricted with restriction endonucleases (Gibco/BRL, Gaithersburg, USA) at the conditions recommended by the manufacturer. All other genetic procedures and modifications of DNA were conducted as described by Sambrook et al. (1989).
Transfer of DNA by electroporation
Plasmids pNC9503, pBBRKmNC903, pDBMCS-2, pDBMCS-5, pCG76, pCGredORF6, pCGredORF42, pCGORF6::aph, and pCGORF6::aacC1 were transferred to G. westfalica strain Kb1 by electroporation using a model 2510 electroporator (Eppendorf-Netheler-Hinz, Hamburg, Germany) according to a previously described method (Arenskötter et al. 2003). Transfer of the isolated megaplasmid pKB1 from G. westfalica strain Kb1 and of the recombinant plasmid pKB1pBBRORF34::aph to species of the genera Gordonia and to M. smegmatis strain mc2155 was performed by electroporation according to the same method. Also, purified and desalted plasmid pBBRORF34::aph was transferred to the pKB1-containing recombinant G. polyisoprenivorans strain VH2 and M. smegmatis strain mc2155 by electroporation. Preparation of electrocompetent cells and execution of electroporation were done as described recently (Kalscheuer et al. 1999; Arenskötter et al. 2003). Plasmid pKB1pBBRORF34::aph was transferred to E. coli strain DH5α by electroporation. Preparation of electrocompetent cells of E. coli strain DH5α and execution of electroporation were performed according to a protocol described by Sheng et al. (1995).
Transfer of DNA by conjugation
Transfer of plasmids pBBRKmNC903, pDBMCS-2, and pDBMCS-5 (Table 1) was performed by conjugation, applying a previously described protocol (Friedrich et al. 1981), using E. coli S17–1 as donor and G. westfalica strain Kb1 as recipient. Conjugative transfer of plasmid pKB1 was carried out by applying the same protocol (Friedrich et al. 1981) using G. westfalica strain Kb1 as donor and mutant strain PD630Km100 of R. opacus as recipient at a 1:1 cell ratio. Plasmid mobilization frequencies were determined by plating appropriate serial dilutions of the mating cell resuspensions onto selective media containing 500 μM CdCl2 and by plating in parallel dilutions of the control recipient culture to obtain recipient viable counts on non-selective media. Mating efficiencies were calculated as transconjugant colony forming units (CFU) per recipient viable cell counts.
Detection of pKB1-harboring transformants
Single colonies of transformed cells were transferred into 5 ml of selective medium (500 μM CdCl2) with toothpicks. The presence of pKB1 in CdCl2-resistant transformants of strains of the genus Gordonia, R. opacus strain PD630Km100, and M. smegmatis strain mc2155 was verified by plasmid isolation. Plasmid DNA was extracted by a modified procedure of the alkaline extraction method (Birnboim and Doly 1979), and the identity of pKB1 was demonstrated by restriction with appropriate endonucleases (Gibco/BRL; Fig. 2).
Gel permeation chromatography
Cleavage of poly(cis-1,4-isoprene) (#182141, Sigma-Aldrich, Steinheim, Germany) by recombinant pKB1-harboring strains of R. opacus PD630Km100 and of M. smegmatis mc2155 was investigated by gel permeation chromatography (GPC). After a cultivation period of 8 weeks, samples were prepared in chloroform and analyzed as previously described (Ibrahim et al. 2006) employing a Waters–GPC system (Waters, Milford, CT, USA), consisting of a 515-HPLC pump, a 410 differential refractometer, a 717plus autosampler, and four in-series-connected Styragel columns (HR3, HR4, HR5, and HR6 with pore sizes of 103, 104, 105, or 106 Å, respectively). Molecular weights of poly(cis-1,4-isoprene) were calculated from retention times of defined poly(cis-1,4-isoprene) standards (PSS Polymer Standards Service GmbH, Mainz, Germany).
Plasmid constructions
Plasmids pCGredORF6 and pCGredORF42 were constructed to test their ability to integrate into plasmid pKB1 in its natural host G. westfalica strain Kb1. Using the two sets of oligonucleotides, P4226fBamHI plus P4863bXbaI and P39530fBamHI plus P40671bXbaI (Table 2), and Pfx DNA polymerase (Gibco BRL) according to the manufacturer’s instructions, 637- or 1,141-bp sequences of ORF6 or ORF42 were amplified by PCR, respectively. The PCR products were cloned into SmaI-linerarized plasmid pBluescript SK− (Stratagene, San Diego, USA), excised by restriction with BamHI and XbaI, and ligated to BamHI–XbaI linearized plasmid pCG76, yielding plasmids pCGredORF6 and pCGredORF42, which were transferred to G. westfalica strain Kb1 by electroporation.
To obtain stable mutants of G. westfalica strain Kb1, a gentamycin or kanamycin resistance gene should be integrated into pKB1 via homologous recombination. Therefore, two 1,469- and 1,430-bp pKB1-specific fragments were amplified by PCR using the two oligonucleotide sets, P3042fBamHI plus P4510bHindIII and P4510fHindIII plus P5940bXbaI (Table 2), and Pfx DNA polymerase (Gibco BRL). The two obtained PCR products were cloned into SmaI-linerarized plasmid pBluescript SK− (Stratagene); the 1,469- and 1,430-bp PCR products were excised by restriction with BamHI plus HindIII or with HindIII plus XbaI, respectively. Both fragments and either an about 1,000-bp HindIII–HindIII kanamycin resistance cassette (aph) or an about 1,000-bp HindIII–HindIII gentamycin resistance cassette (aacC1) were ligated to BamHI–XbaI-linearized plasmid pCG76, yielding plasmids pCGORF6::aph and pCGORF6::aacC1, which were transferred to E. coli strain XL1-Blue. Kanamycin- or gentamycin-resistant transformants harboring pCGORF6::aph or pCGORF6::aacC1 were obtained, respectively. These plasmids were then isolated and transferred to G. westfalica strain Kb1 by electroporation.
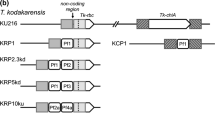
For construction of the suicide vector pBBRORF34::aph, the 2,192-bp sequence of the cadA coding region of plasmid pKB1 (position 29,826–32,018) exhibited a unique restriction site for Eco72I 1,711 nucleotides downstream of the start codon. The entire 2,192-bp cadA region was amplified by PCR using oligonucleotides P29826fBamHI and P32018bXbaI (Table 2). The PCR product was then cloned into SmaI-linearized plasmid pBluescript SK− (Stratagene), excised by restriction with BamHI and XbaI, ligated to BamHI–XbaI-linearized plasmid pBBR1MCS-5 (Kovach et al. 1995) and transferred into E. coli strain XL1-Blue. Gentamycin-resistant colonies were obtained, and hybrid plasmid pBBRORF34 was isolated and sequenced. Because plasmid pBBR1MCS-5 contains no cleavage site for Eco72I, pBBRORF34 was linearized by restriction with Eco72I at position 1,711 of cadA. An about 1,000-bp SmaI–SmaI kanamycin resistance cassette (aph) was subsequently inserted at this position of cadA, yielding vector pBBRORF34::aph, which was transferred to E. coli strain XL1-Blue. Gentamycin- and kanamycin-resistant colonies were obtained. The final vector pBBRORF34::aph was sequenced and contained a kanamycin-resistance and a gentamycin-resistance gene as selectable markers. Because pBBRORF34::aph lacked an origin of replication for Gordonia and Mycobacterium, it represented a suicide plasmid unable to replicate in these bacteria. Construction of this plasmid is summarized in Fig. 1.

Scheme for the construction of plasmid pBBRORF34::aph derived from pBBR1MCS-5. Relevant cleavage sites and structural genes are indicated (aacC1, gentamycin-3-acetyltransferase gene or gentamycin resistance gene; aph, aminoglycoside phosphotransferase gene or kanamycin resistance gene; lacZ, β-galactosidase gene of E. coli; rep, E. coli origin of replication; mob, gene for plasmid mobilization when RK2 transfer functions are provided in trans; cadA, cadmium resistance gene of plasmid pKB1)
Nucleotide sequence analysis
The cadA containing hybrid plasmids pBBRORF34 and pBBRORF34::aph were sequenced, employing IRD800-labeled universal and reverse primers, the SequiTherm Excel II Long-Read L-C kit, and a LI-COR 4200 sequencer (LI-COR Biosiences, Lincoln, Nebr.).
Results
Failure to transfer plasmids stably into G. westfalica strain Kb1
Transformation of G. westfalica strain Kb1 and, therefore, engineering of pKB1 failed because of the absence of efficient gene transfer systems for this strain. It was, for example, not possible to transfer plasmids like pNC9503 or pBBRKmNC903 (Arenskötter et al. 2003) and pDBMCS-2 or pDBMCS-5 (Bröker et al. 2004), suitable for other members of the genus Gordonia, to G. westfalica strain Kb1 by electroporation or by conjugation via E. coli strain S17-1.
Because the thermosensitive plasmid pCG79, a derivative of plasmid pCG76 containing Tn611 for transposition, was able to replicate autonomously in G. polyisoprenivorans strain VH2 (Banh et al. 2005), the E. coli–mycobacteria shuttle vector pCG76 was transferred to G. westfalica strain Kb1 by electroporation. Approximately 80% of the obtained transformants, which were selected on St-I agar plates containing streptomycin (20 μg/ml), harbored plasmid pCG76, indicating autonomous replication of this plasmid in G. westfalica strain Kb1 at 30°C.
The thermosensitive plasmid pCG76 maintains in cells incubated at 30°C but is rapidly lost when the culture is incubated at 39°C (Guilhot et al. 1994). As G. westfalica strain Kb1 is able to grow until temperatures of 42°C, plasmid pCG76 can be applied as suicide plasmid in this strain and used for plasmid integration via homologous recombination between a pKB1-specific sequence on plasmid pCG76 and the respective sequence in pKB1. As it was previously discussed that the putative gene products of the pKB1-encoded ORF6 (putative epoxide hydrolase) and ORF42 (putative cytochrome c oxidase) might have a metabolic function (Bröker et al. 2004), internal regions of ORF6 and ORF42 were used as pKB1-specific sequences to test the possibility to disrupt these ORFs by the aspired plasmid integration. Therefore, 637- and 1,141-bp sequences of ORF6 or ORF42, respectively, of plasmid pKB1 were amplified by PCR and ligated to plasmid pCG76, yielding plasmids pCGredORF6 and pCGredORF42, which were then transferred to G. westfalica strain Kb1 by electroporation. Selection of mutants with an integration of plasmid pCGredORF6 or pCGredORF42 and the required temperature shift to 40°C was performed as previously described (Guilhot et al. 1994; Banh et al. 2005). Plasmid DNA was isolated from 50 obtained streptomycin-resistant mutants. Only two mutants still harbored beside pKB1 episomal pCG76 derivative DNA, indicating loss or integration of pCGredORF6 or pCGredORF42, respectively, in the other mutants. Plasmid DNA isolated from the wild-type G. westfalica strain Kb1 and from ten mutants with putative integration of pCGredORF6 (designated ORF6M1 to ORF6M10) or pCGredORF42 (designated ORF42M1 to ORF42M10), respectively, were restricted with appropriate restriction endonucleases and separated in a 0.7% (w/v) agarose gel, which was stained with ethidium bromide. Comparison of the visualized restriction fragment pattern of plasmid DNA derived from the mutants to that of the wild type revealed the occurrence of an additional fragment with an increased size of 9.9 or 10.4 kbp, representing the size of vector pCGredORF6 or pCGredORF42, respectively, for the mutants. These results demonstrated integration of pCGredORF6 or pCGredORF42 into plasmid pKB1.
Mutants with an integration of pCG76 into plasmid pKB1 were further analyzed with respect to their capabilities to utilize rubber. Therefore, the two mutants ORF6M9 and ORF42M6 and also the wild-type G. westfalica strain Kb1 were exemplarily cultivated with poly(cis-1,4-isoprene) as the only carbon source at 40°C to prevent excision of the integrated thermosensitive vector pCG76. However, G. westfalica strain Kb1 wild type showed only very slow growth at 40°C with poly(cis-1,4-isoprene) as sole carbon source, which was also observed for G. polyisoprenivorans strain VH2 (Banh et al. 2005). Another problem was the acquired resistance to streptomycin of G. westfalica strain Kb1. In spite of selection with streptomycin (20 μg/ml), mutants ORF6M9 and ORF42M6 reverted to the wild-type genotype after two generations with a complete loss of the integrated plasmids pCGredORF6 or pCGredORF42, respectively (data not shown).
To overcome these problems, the integration of a gentamycin (aacC1) or kanamycin (aph) resistance gene into plasmid pKB1 via homologous recombination was targeted. Therefore, two pKB1-specific PCR products and a kanamycin or a gentamycin resistance cassette were ligated to pCG76, yielding plasmids pCGORF6::aph and pCGORF6::aacC1, respectively. After transfer of these hybrid plasmids to G. westfalica strain Kb1 by electroporation, neither kanamycin- nor gentamycin-resistant transformants were obtained, indicating that the plasmid-encoded resistance genes were not functionally expressed in G. westfalica strain Kb1. Therefore, direct genetic manipulation of plasmid pKB1 in its natural host G. westfalica strain Kb1 failed. Instead, megaplasmid pKB1 had to be transferred to genetically approachable strains to engineer pKB1 in the resulting transgenic strains.
Transfer of megaplasmid pKB1 by electroporation and transformation efficiency
From a previous study, it was known that G. westfalica strain Kb1 harboring megaplasmid pKB1 is able to grow in the presence of up to 800 μM CdCl2 on St-I agar plates, whereas a mutant cured of this plasmid exhibited decreased cadmium resistance (Bröker et al. 2004). Tolerance for cadmium was investigated and evaluated after incubation for 3 days at 30°C on St-I agar plates containing CdCl2 at concentrations between 100 and 800 μM for some other related bacteria. It was found that G. polyisoprenivorans strains VH2 (growth up to 150 μM cadmium) and Y2K (growth up to 150 μM cadmium) or M. smegmatis strain mc2155 (growth up to 100 μM cadmium) and R. opacus strain PD630Km100 (growth up to 100 μM cadmium) exhibited a significantly lower cadmium resistance (Table 3). When electrocompetent cells of these three cadmium-sensitive strains were transformed with isolated megaplasmid pKB1 by electroporation, CdCl2-resistant colonies were obtained on plates containing 500 μM CdCl2, whereas no colonies occurred on the same plates with cells subjected to electroporation without plasmid. Transformation frequencies of plasmid pKB1 were 5.5 × 103, 1.9 × 103, and 8.3 × 102 transformants per µg plasmid DNA for G. polyisoprenivorans strain VH2, G. polyisoprenivorans strain Y2K, and M. smegmatis strain mc2155, respectively, with each value representing the average from three independent experiments. Presence of megaplasmid pKB1 in the CdCl2-resistant transformants was confirmed by isolation of plasmid DNA from cells; in addition, the restriction fragment patterns of these plasmids were identical with that of the plasmid isolated from the wild type (Fig. 2). Therefore, the pKB1-encoded cadmium resistance gene cadA could be used as selection marker.

Comparison of the restriction pattern of plasmid pKB1 from the native host versus the heterologous hosts. The isolated plasmid pKB1 DNA of the native host G. westfalica strain Kb1 and of the heterologous hosts R. opacus strain PD630Km100, M. smegmatis strain mc2155, and G. polyisoprenivorans strains Y2K and VH2 was digested with BamHI, separated by agarose gel electrophoresis and stained with ethidium bromide. M λ-DNA digested with PstI, 1 pKB1 of G. westfalica strain Kb1, 2 pKB1 of the recombinant R. opacus strain PD630Km100, 3 pKB1 of the recombinant M. smegmatis strain mc2155, 4 pKB1 of the recombinant G. polyisoprenivorans strain Y2K, and 5 pKB1 of the recombinant G. polyisoprenivorans strain VH2
Transfer of pKB1 by conjugation and megaplasmid mobilization frequency
To determine, whether megaplasmid pKB1 is transmissible by conjugation, a spontaneous kanamycin-resistant mutant of R. opacus strain PD630, which was referred to as PD630Km100, was generated and used as recipient for conjugative transfer of megaplasmid pKB1 by spot agar mating with G. westfalica strain Kb1 as donor. For selection of transconjugants from recipient and donor cells, the pKB1-encoded cadmium resistance gene cadA was used as selection marker. Colonies of transconjugants of R. opacus strain PD630Km100 appeared after 4 days of incubation on St-I agar plates containing 100 μg/ml kanamycin and 500 μM CdCl2. Mobilization frequencies of megaplasmid pKB1 to the recipient R. opacus strain PD630Km100 were approximately 6.2 × 10−8 events per recipient cell as revealed in three independent experiments. Presence of plasmid pKB1 in the transconjugants was also confirmed physically by electrophoresis in crude lysates (Fig. 2).
Characterization of the recombinant plasmid pKB1-containing strains
Plasmid pKB1 conferred cadmium resistance to G. polyisoprenivorans strains VH2 and Y2K, as well as to M. smegmatis strain mc2155 and R. opacus strain PD630Km100. In contrast to the parent strains, all recombinant strains of these bacteria harboring pKB1 were able to grow on St-I agar plates containing the maximum concentration of 800 μM CdCl2 (Table 3).
To investigate whether plasmid pKB1 confers also poly(cis-1,4-isoprene) utilization to the non-rubber-degrading bacteria M. smegmatis strain mc2155 and R. opacus strain PD630Km100, the recombinant pKB1-harboring strains were cultivated in MSM containing 0.25% (w/v) poly(cis-1,4-isoprene) as sole carbon and energy source. However, even after 30 days incubation the recombinant pKB1-containing strains did not exhibit any growth on or deterioration of poly(cis-1,4-isoprene). GPC analysis of the residual poly(cis-1,4-isoprene) with an average molecular mass of 800-kDa after 8 weeks of incubation did not indicate any changes in molecular weights or concentrations of the polymer (data not shown).
Modification of plasmid pKB1
The recombinant pKB1-harboring G. polyisoprenivorans strain VH2 and M. smegmatis strain mc2155 were employed to modify pKB1. After amplification of cadA from G. westfalica strain Kb1 by PCR using pKB1 as template and primers P29826fBamHI and P32018bXbaI, the PCR product was restricted with BamHI plus XbaI and then ligated to BamHI–XbaI-linearized plasmid pBBR1MCS-5 DNA, yielding plasmid pBBRORF34 as described in “Materials and methods.” After restriction of pBBRORF34 with Eco72I, the linearized plasmid pBBRORF34 was ligated with a SmaI-restricted kanamycin resistance cassette (aph) to generate the vector pBBRORF34::aph (Fig. 1).
To examine the ability of the suicide plasmid pBBRORF34::aph to integrate into plasmid pKB1, the purified and desalted plasmid pBBRORF34::aph was transferred to the pKB1-harboring G. polyisoprenivorans strain VH2 and M. smegmatis strain mc2155 by electroporation. Selection of kanamycin- (50 μg/ml) and gentamycin (10 μg/ml)-resistant transformants yielded recombinant strains in which pBBRORF34::aph had integrated into the target locus on plasmid pKB1 by a single-crossover event after homologous recombination. In total, 34 recombinant strains of M. smegmatis strain mc2155, designated M1–M34, and 11 recombinant strains of G. polyisoprenivorans strain VH2, designated G1–G11, were obtained, which harbored the modified plasmid pKB1 with inserted vector pBBRORF34::aph and designated as pKB1pBBRORF34::aph. All 45 transformants harboring the modified plasmid pKB1pBBRORF34::aph were verified by isolation of plasmid DNA from kanamycin- and gentamycin-resistant cells and by PCR using primers P29826fBamHI and P32018bXbaI. This gave two products: one 2,192-bp fragment representing the intact cadA gene and one 3,192-bp representing cadA with the inserted 1,000-bp kanamycin resistance cassette (data not shown). Therefore, plasmid pKB1pBBRORF34::aph resulted from a single-crossover event of pBBRORF34::aph into pKB1 with one wild-type copy of cadA plus the vector copy containing the inserted kanamycin resistance cassette, as it was expected for heterogenotes occurring after the performed selection of kanamycin and gentamycin. Thus, plasmid pKB1 was provided with these two additional antibiotic selection markers. If plasmid pKB1pBBRORF34::aph from transformants M1 and G1 was isolated and transferred to G. polyisoprenivorans strains VH2 and Y2K, and to M. smegmatis strain mc2155 by electroporation, kanamycin- and gentamycin-resistant transformants were obtained from either strain, thus, indicating a stable propagation of plasmid pKB1pBBRORF34::aph in these strains. The presence of this plasmid was confirmed as described above.
Unfortunately, transfer of pKB1pBBRORF34::aph to G. alkanivorans strain 44369 by electroporation failed; no kanamycin- (50 μg/ml) and gentamycin (10 μg/ml)-resistant transformants were obtained. Plasmid pKB1pBBRORF34::aph, which should contain an origin of replication for E. coli because of the integration of vector pBBRORF34::aph, was also transferred to E. coli strain DH5α by electroporation according to a protocol described by Sheng et al. (1995). Although strain DH5α was described to be more efficient for transformation of large DNA molecules (Sheng et al. 1995), neither gentamycin- nor kanamycin-resistant transformants were obtained after transformation.
Discussion
Previous investigations provided evidence that genes essential for rubber degradation in G. westfalica strain Kb1 are encoded on megaplasmid pKB1 (Bröker et al. 2004). As genetic engineering of pKB1 in its host failed because of the absence of an effective gene transfer system for this strain, pKB1 had to be transferred to genetically approachable strains for engineering. The suitability of the cadmium resistance gene cadA (ORF34) as pKB1-specific selection marker to monitor transfer of pKB1 by electroporation and conjugation to two cadmium sensitive strains of G. polyisoprenivorans and two taxonomically related bacteria was unequivocally demonstrated with pKB1 mediating cadmium resistance to a concentration of 800 μM (Table 3).
Transfer of megaplasmid pKB1 by electroporation occurred at transformation frequencies of 5.5 × 103 and 1.9 × 103 transformants per microgram plasmid DNA for G. polyisoprenivorans strains VH2 and Y2K, and 8.3 × 102 for M. smegmatis strain mc2155, respectively. It is described that efficiencies of plasmid DNA transfer by electroporation decrease with increasing plasmid size (Mozo and Hooykaas 1991), which is consistent with transformation frequencies for the 101,016-bp plasmid pKB1.
Conjugative transfer of megaplasmid pKB1, using the spontaneous kanamycin-resistant mutant R. opacus strain PD630Km100 as recipient and G. westfalica strain Kb1 as donor occurred at mobilization frequencies of approximately 6.2 × 10−8 events per recipient cell, demonstrating the functionality of pKB1-encoded conjugative transfer genes. Therefore, plasmid pKB1 represents the first conjugative plasmid within the genus Gordonia. Strains of the genus Rhodococcus are the closest relatives containing described conjugative megaplasmids, as for example, megaplasmids pREA400 (400 kbp) and pREA250 (250 kbp) from R. erythropolis strain AN12, which were shown to be conjugative for the recipient R. erythropolis strain SQ1 at frequencies of approximately 7 × 10−4 events per recipient cell (Yang et al. 2007). Conjugative transfer of plasmid pD188 (138 kbp) from R. fascians strain D188 between different R. fascians strains occurred at transfer frequencies of 1.5 × 10−2 to 3 × 10−7 (Desomer et al. 1988).
Plasmid pKB1 did not confer the utilization of poly(cis-1,4-isoprene) to the non-rubber-degrading bacteria M. smegmatis strain mc2155 and R. opacus strain PD630Km100 as revealed by the lacking growth and by the GPC analysis of the residual poly(cis-1,4-isoprene) after 8 weeks of incubation. It is, therefore, suggested that plasmid pKB1-encoded genes are not essential for the initial poly(cis-1,4-isoprene) cleavage but code for enzymes required for subsequent steps of the catabolic pathway. Because the rubber-degrading strain G. alkanivorans strain 44187 also harbors a megaplasmid-designated pKB2, which consists of plasmid pKB1-identical sequence regions (Bröker et al., unpublished data), and because the difference between this and the non-rubber-utilizing strain 44369 of G. alkanivorans seems to be only the absence of plasmid pKB2 (Kummer et al. 1999), plasmid pKB1 should be transferred to strain 44369 to restore the ability for rubber degradation. Because of the cadmium resistance of G. alkanivorans strain 44369, plasmid pKB1 had to be provided with other resistance markers.
Thus, two additional antibiotic selection markers (kanamycin and gentamycin resistance) were accommodated in pKB1 to facilitate transfer of the plasmid into cadmium-sensitive strains of Gordonia and related taxa for analysis of plasmid encoded features. As transfer of foreign DNA to G. westfalica strain Kb1 failed, the genetically approachable recombinant pKB1-containing G. polyisoprenivorans strain VH2 and M. smegmatis strain mc2155 were used for engineering of pKB1. Methods for transformation and gene replacement were intensively studied for M. smegmatis strain mc2155 (Husson et al. 1990; McFadden 1996; Hinds et al. 1999; Galamba et al. 2001) and for G. polyisoprenivorans strain VH2 (Arenskötter et al. 2003; Banh et al. 2005; Arenskötter et al., unpublished data). Heterogenotes, containing a modified plasmid pKB1, in which the suicide vector pBBRORF34::aph (Fig. 1) integrated into pKB1 by homologous recombination, were obtained for both recombinant strains. The modified plasmid pKB1, designated as pKB1pBBRORF34::aph, contained two additional useful antibiotic selection markers (kanamycin and gentamycin resistance) and mediated resistance to both antibiotics to recombinant pKB1pBBRORF34::aph containing strains of G. polyisoprenivorans strains VH2 and Y2K and M. smegmatis strain mc2155 but unfortunately not to G. alkanivorans strain 44369 representing one of the closest relative of G. westfalica strain Kb1 (Arenskötter et al. 2005). It was suggested that plasmid-encoded kanamycin or gentamycin resistance genes were not functionally expressed neither in G. alkanivorans strain 44369 using plasmid pKB1pBBRORF34::aph nor in G. westfalica strain Kb1 using plasmids pCGORF6::aph and pCGORF6::aacC1. Also transfer and propagation of foreign DNA seemed to be prevented by these members of the genus Gordonia. Possibly, these strains possess a highly efficient protection system against foreign DNA, such as restriction–modification (R–M) systems (Wilson and Murray 1991), a histone-like nucleoid structure-like (H-NS) protein repression system (Navarre et al. 2007) or a putative RNA-interference-based immune system (Makarova et al. 2006), as it has been described or discussed for many other bacteria.
As there is obviously a lack of suitable genetic tools for Gordonia species, the procedures applied in this study open the possibility for transfer of large DNA molecules by conjugation and electroporation to genetically approachable strains and for their engineering in resulting transgenic strains.
References
Arenskötter M, Baumeister D, Berekaa MM, Pötter G, Kroppenstedt RM, Linos A, Steinbüchel A (2001) Taxonomic characterization of two rubber-degrading bacteria belonging to the species Gordonia polyisoprenivorans and analysis of hypervariable regions of 16S rDNA sequences. FEMS Microbiol Lett 205:277–282
Arenskötter M, Baumeister D, Kalscheuer R, Steinbüchel A (2003) Identification and application of plasmids suitable for transfer of foreign DNA to members of the genus Gordonia. Appl Environ Microbiol 69:4971–4974
Arenskötter M, Bröker D, Steinbüchel A (2004) Biology of the metabolically diverse genus Gordonia. Appl Environ Microbiol 70:3195–3204
Arenskötter M, Linos A, Schumann P, Kroppenstedt RM, Steinbüchel A (2005) Gordonia nitida Yoon et al. 2001 is a later synonym of Gordonia alkanivorans Kummer et al. 1999. Int J Syst Evol Microbiol 55:695–697
Banh Q, Arenskötter M, Steinbüchel A (2005) Establishment of Tn5096-based transposon mutagenesis in Gordonia polyisoprenivorans. Appl Environ Microbiol 71:5077–5084
Birnboim HC, Doly J (1979) A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7:1513–1523
Bröker D, Arenskötter M, Legatzki A, Nies DH, Steinbüchel A (2004) Characterization of the 101.016-kbp megaplasmid pKB1 isolated from the rubber degrading bacterium Gordonia westfalica Kb1. J Bacteriol 186:212–225
Bullock WO, Fernandez JM, Stuart JM (1987) XL1-Blue: a high efficiency plasmid transforming recA Escherichia coli strain with β-galactosidase selection. Bio Techniques 5:376–379
Byrd DR, Matson SW (1997) Nicking by transesterification: the reaction catalysed by a relaxase. Mol Microbiol 25:1011–1022
Desomer J, Dhaese P, van Montagu M (1988) Conjugative transfer of cadmium resistance plasmids in Rhodococcus fascians strains. J Bacteriol 170:2401–2405
Dogan I, Pagilla KR, Webster DA, Stark BC (2006) Expression of Vitreoscilla hemoglobin in Gordonia amarae enhances biosurfactant production. J Ind Microbiol Biotechnol 33:693–700
Friedrich B, Hogrefe C, Schlegel HG (1981) Naturally occurring genetic transfer of hydrogen-oxidizing ability between strains of Alcaligenes eutrophus. J Bacteriol 147:198–205
Galamba A, Soetaert K, Wang X-M, De Bruyn J, Jacobs P, Content J (2001) Disruption of adhC reveals a large duplication in the Mycobacterium smegmatis mc2155 genome. Microbiology 147:3281–3294
Gilbert SC, Morton J, Buchanan S, Oldfield C, McRoberts A (1998) Isolation of a unique benzothiophene-desulphurizing bacterium, Gordona sp. strain 213E (NCIMB 40816), and characterization of desulphurization pathway. Microbiology (SGM) 144:2545–2553
Grohmann E, Muth G, Espinosa M (2003) Conjugative plasmid transfer in Gram-positive bacteria. Microbiol Mol Biol Rev 67:277–301
Guilhot C, Otal I, van Rompaey I, Martìn C, Gicquel B (1994) Efficient transposition in mycobacteria: construction of Mycobacterium smegmatis insertional mutant libraries. J Bacteriol 176:535–539
Hernandez-Perez G, Fayolle F, Vandecasteele J-P (2001) Biodegradation of ethyl t-butyl ether (ETBE), methyl t-butyl ether (MTBE) and t-amyl methyl ether (TAME) by Gordonia terrae. Appl Microbiol Biotechnol 55:117–121
Hinds J, Mahenthiralingam E, Kempsell KE, Duncan K, Stokes RW, Parish T, Stoker NG (1999) Enhanced gene replacement in mycobacteria. Microbiology 145:519–527
Husson RN, James BE, Young RA (1990) Gene replacement and expression of foreign DNA in mycobacteria. J Bacteriol 172:519–524
Ibrahim EMA, Arenskötter M, Luftmann H, Steinbüchel A (2006) Identification of poly(cis-1,4-isoprene) degradation intermediates during growth of moderately thermophilic Actinomycetes on rubber and cloning of a functional lcp homologue from Nocardia farcinica strain E1. Appl Environ Microbiol 72:3375–3382
Kalscheuer R, Arenskötter M, Steinbüchel A (1999) Establishment of a gene transfer system for Rhodococcus opacus PD630 based on electroporation and its application for recombinant biosynthesis of poly(3-hydroxyalkanoic acids). Appl Microbiol Biotech 52:508–515
Kim SB, Brown R, Oldfield C, Gilbert SC, Goodfellow M (1999) Gordonia desulfuricans sp. nov., a benzothiophene-desulfurizing actinomycete. Int J Syst Bacteriol 49:1845–1851
Kim SB, Brown R, Oldfield C, Gilbert SC, Iliarionov S, Goodfellow M (2000) Gordonia amicalis sp. nov., a dibenzothiophene–desulfurizing actinomycete. Int J Syst Bacteriol 50:2031–2036
Kotani T, Yamamoto T, Yurimoto H, Sakai Y, Kato N (2003) Propane monooxygenase and NAD+-dependent secondary alcohol dehydrogenase in propane metabolism by Gordonia sp. strain TY-5. J Bacteriol 185:7120–7128
Kotani T, Yurimoto H, Kato N, Sakai Y (2007) Novel acetone metabolism in a propane-utilizing bacterium, Gordonia sp. strain TY-5. J Bacteriol 189:886–893
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM (1995) Four new derivates of the broad host range cloning vector pBBR1MCS, carrying different antibiotic resistance cassettes. Gene 166:175–176
Kummer C, Schumann P, Stackebrandt E (1999) Gordonia alkanivorans sp. nov., isolated from tar-contaminated soil. Int J Syst Bacteriol 49:1513–1522
Linos A, Steinbüchel A (1998) Microbial degradation of natural and synthetic rubbers by novel bacteria belonging to the genus Gordona. Kautsch Gummi Kunstst 51:496–499
Linos A, Steinbüchel A, Spröer C, Kroppenstedt RM (1999) Gordonia polyisoprenivorans sp. nov., a rubber degrading actinomycete isolated from automobile tire. Int J Syst Bacteriol 49:1785–1791
Linos A, Berekaa MM, Steinbüchel A, Kim KK, Spröer C, Kroppenstedt RM (2002) Gordonia westfalica sp. nov., a novel rubber-degrading actinomycete. Int J Syst Evol Microbiol 52:1133–1139
Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1:7–32
Mozo T, Hooykaas PJJ (1991) Electroporation of megaplasmids into Agrobacterium. Plant Mol Biol 16:917–918
McFadden J (1996) Recombination in mycobacteria. Mol Microbiol 21:2056–211
Navarre WW, McClelland M, Libby SJ, Fang FC (2007) Silencing of xenogeneic DNA by H-NS—facilitation of lateral gene transfer in bacteria by a defense system that recognizes foreign DNA. Genes Dev 21:1456–1471
Pansegrau W, Lanka E (1996) Enzymology of DNA transfer by conjugative mechanisms. Prog Nucleic Acid Res Mol Biol 54:197–251
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Schlegel HG, Kaltwasser H, Gottschalk G (1961) Ein Submersverfahren zur Kultur wasserstoffoxidierender Bakterien: Wachstumsphysiologische Untersuchungen. Arch Mikrobiol 38:209–222
Sheng Y, Mancino V, Birren B (1995) Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res 23:1990–1996
Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR Jr (1990) Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4:1911–1919
Wilson GG, Murray NE (1991) Restriction and modification systems. Annu Rev Genet 25:585–627
Yang JC, Lessard PA, Sengupta N, Windsor SD, O’Brien XM, Bramucci M, Tomb J-F, Nagarajan V, Sinskey AJ (2007) TraA is required for megaplasmid conjugation in Rhodococcus erythropolis AN12. Plasmid 57:55–70
Veiga-Crespo P, Feijoo-Siota L, de Miguel T, Poza M, Villa TG (2006) Proposal of a method for the genetic transformation of Gordonia jacobae. J Appl Microbiol 100:608–614
Yoon J-H, Lee JJ, Kang SS, Takeuchi M, Shin YK, Lee ST, Kang KH, Park YH (2000) Gordonia nitida sp. nov., a bacterium that degrades 3-ethylpyridine and 3-methylpyridine. Int J Syst Evol Microbiol 50:1203–1210
Acknowledgment
Financial support by a grant of the Deutsche Forschungsgemeinschaft (STE 386/10–1) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bröker, D., Arenskötter, M. & Steinbüchel, A. Transfer of megaplasmid pKB1 from the rubber-degrading bacterium Gordonia westfalica strain Kb1 to related bacteria and its modification. Appl Microbiol Biotechnol 77, 1317–1327 (2008). https://doi.org/10.1007/s00253-007-1262-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-1262-8