
Abstract
From the culture filtrate of the fungus Dactylellina varietas (syn. Dactylella varietas), an extracellular protease (designed Dv1) was purified by cation exchange and hydrophobic interaction chromatography. The purified protease showed a molecular mass of approximately 30 kDa and displayed an optimal activity at pH 8 and 60.5°C (more than 20 min). This protease could degrade a broad range of substrates including casein, gelatin, BSA (bovine serum albumin), and nematode cuticle. However, its proteolytic activity was highly sensitive to the serine protease inhibitor Phenylmethylphonylfuoride (1 mM), indicating that it belongs to the serine-type peptidase group. This protease could immobilize the free-living nematodes Panagrellus redivivus and Caenorhabditis elegans and hydrolyze the purified cuticle of P. redivivus, suggesting it may play a role in infection against nematodes. The encoding gene of Dv1 and its promoter sequence were cloned using degenerate primers and the DNA walking technology. Its open-reading frame contains 1,224 base pairs and without any intron. The deduced amino-acid sequence shared low identity to serine proteases from other nematode-trapping fungi. Our report identified a novel pathogenic protease from the nematode-trapping fungus D. varietas, and the three-dimensional structure of this protease was predicted using the Swiss-Prot method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant-parasitic nematodes have been reported to cause severe damages to agriculture all over the world (Huang et al. 2004). Chemical pesticides have been the predominant agents for controlling these pests. However, chemical pesticides can cause a series of environmental problems. Nematode-trapping fungi, natural enemies of nematodes, have been suggested as potential biocontrol agents to control the harmful nematodes (Siddiqui and Mahmood 1996). Infection of nematodes by nematode-trapping fungi proceeds first by adhering to and trapping of the nematode. This is then followed by penetration, immobilization, and finally, digestion of the nematode (Jansson and Nordbring-Hertz 1988; Tunlid and Jansson 1991; Yang et al. 2005a). The pathogenesis of nematode-trapping fungi has been assumed to be due to the combination of mechanical forces (capturing devices) and hydrolytic enzymes (e.g., serine protease; Tunlid et al. 1994; Wang et al. 2006; Yang et al. 2005a).
The nematode cuticle is a thin and flexible exo-skeleton, composed primarily of proteins including collagens (Cox et al. 1981; Maizels et al. 1993). Extracellular enzymes, including serine protease, chitinase and collagenase, could digest the main chemical constituents of the nematode cuticle and eggshell, and these enzymes have been reported to be involved in the infectious process (Huang et al. 2004). Since Lopez-Llorca (1990) isolated the first serine protease from the endoparasitic fungus Pochonia rubescens (syn. Verticillium suchlasporium), several other serine proteases have been identified from other nematophagous fungi (e.g., Bonants et al. 1995; Segers et al. 1994; Tunlid et al. 1994; Yang et al. 2005a). Among them, two were identified from the nematode-trapping fungi Arthrobotrys oligospora (PII; Tunlid et al. 1994) and Monacrosporium microscaphoides (Wang et al. 2006). These two fungi can immobilize nematodes through their adhesive nets. At present, little is known about the pathogenic proteases from other nematode-trapping fungi.
Dactylella Grove is an important genus, containing several nematode-trapping species. This genus included nonpredatory species as well as predatory species (Subramanian 1963). In our previous report, we identified that species Dactylellina varietas (Dactylella varietas) produced two kinds of capturing devices: adhesive knobs and nonconstricting rings. It was reported as a new predatory species by Li et al. (2006).
We recently demonstrated that the nematode-trapping fungus D. varietas produced extracellular enzymes when grown in liquid medium. In this study, an extracellular serine protease present in the culture filtrates of D. varietas was purified and characterized. The purified protease could immobilize the free-living nematodes Panagrellus redivivus and Caenorhabditis elegans and hydrolyze the cuticle proteins of P. redivivus. Interestingly, the encoding gene of Dv1 lacked intron different from other published protease encoding genes of nematophagous fungi.
Materials and methods
Microorganisms and culture conditions
D. varietas (YMF1.00118) was isolated from a soil samples in Yunnan province and had been deposited at the Chinese General Microbiological Culture Collection Center (CGMCC1521). It was maintained on PDA (Potato Dextrose Agar) medium. The PL-4 liquid medium for protease production was described by Yang et al. (2005a). Escherichia coli strain DH5α was used in all DNA manipulations, and this strain was typically grown in Luria-Bertani medium (Yang et al. 2005b) at 37°C.
The free-living nematodes P. redivivus and C. elegans were maintained on oat medium (oat, 20 g; water, 80 ml). Nematodes were separated and washed thoroughly with 50 mM sodium phosphate (pH 7.0) before being used in the assays (Cox et al. 1981; Zhao et al. 2004).
Buffers
The buffers used for protease purification were as follows: buffer A, 10 mM sodium phosphate (pH 6.0); buffer B, 10 mM sodium phosphate (pH 6.0) containing 0.5 M NaCl; buffer C, 50 mM sodium phosphate (pH 7.0) containing 1 M ammonium sulfate; and Buffer D, 50 mM sodium phosphate (pH 7.0). The Britton-Robinson universal buffers (pH 2–12) were used to determine the effects of the pH on enzyme activity (Yang et al. 2005a).
Protease activity analysis
The protease activities of the culture filtrate and purified fractions were qualitatively analyzed by using the casein-plate method as described by Zhao et al. (2004). Quantitative analysis of protease activity was determined by a caseinolytic method described by Wang et al. (2006). Protein concentration was determined by the method of Bradford (1976) using BSA as a standard.
Protease production and purification
D. varietas was cultured in PL-4 liquid medium for 7 days at 26°C. The culture filtrate was collected by vacuum filtration and protease in the filtrate was concentrated by ultrafiltration (5 kDa cutoff membrane, Millipore). The sample was applied to a HiTrap SP FF column (Amersham, Sweden) equilibrated with buffer A, and the bound proteins were eluted with buffer B at 1 ml/min. Fractions containing protease activity from the HiTrap SP FF were pooled and mixed with 3.4 M (NH4)2SO4 in a proportion of 3:2 (v/v, sample: buffer). The sample was applied to a HiPrep 16/10 Phenyl FF (high sub) column (Amersham, Sweden) equilibrated with buffer C, then eluted with buffer D at 2 ml/min. Fractions of 0.5 ml were collected and qualitatively assayed for protease activity. After sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analyses, fractions with protease activity were pooled and stored in −20°C for further analysis.
SDS-PAGE and N-terminal amino-acid sequence analysis
SDS-PAGE was performed with a Mini-PROTEAN III gel system (Bio-Rad, USA), using slab gels (0.5 mm thick, 12% polyacrylamide) according to the method of Laemmli (1970), and the proteins were stained with Coomassie Blue G-250. The N-terminal amino-acid sequence of the purified protease Dv1 was determined according to a previously described method (Wang et al. 2006).
Effect of metal ions and protease inhibitors on the enzyme activity
The purified protease was mixed with its substrate (casein), with different metal ion solutions (0.5 mM) or protease inhibitors (ethylenediamine tetraacetic acid [EDTA], phenylmethylphonylfuoride [PMSF], aprotinins, pepstatin and leupeptin) added, respectively. These mixtures were incubated at optimum reaction condition for 20 min. Their protease activities were then measured according to the method described by Wang et al. (2006). The experiment was repeated three times.
Effects of temperature and pH on enzyme activity
The optimum pH and optimum temperature for enzyme activity as well as the stabilities of the enzyme at various pH and temperatures were determined as described by Yang et al. (2005a). Briefly, the optimum pH was determined by mixing the purified protease with the Britton-Robinson buffer system at pH values between 3 and 9, and the optimum temperature was determined by incubating the reaction mixture at different temperatures ranging from 35 to 75°C.
Nematicidal activity and hydrolysis of protein substrates
The effect of protease on nematodes was investigated by in vitro assays as follows: approximately 50 nematodes were added to solutions of the purified protease Dv1, the crude enzyme solution, and a negative control sample, respectively. The mixtures were incubated at 26°C for 12–24 h, and the numbers of dead nematodes were counted under a light microscope. The experiment was repeated three times.
The purified protease was incubated with different protein substrates at pH 8 and 60.5°C for 20 min and the protease activity was quantitatively assayed. Nematode cuticle was extracted according to the method of Cox et al. (1981).
Gene cloning and sequencing
D. varietas was cultured in PL-4 liquid medium for 4 days at 26°C. Mycelium for DNA extraction was collected by filtration in a sterilized filter funnel and ground to a fine powder in liquid N2. DNA was extracted according to the CTAB method described by Zhang et al. (1996).
A pair of degenerate primers (PF: 5′-AA(A/G)TA(C/T)AT(C/T)GTCGTC(C/T)(A/T)(C/G)AAG-3′; PR: 5′-TTAAG(C/T)(A/G)(G/T)(A/C/T)(G/T)CC(A/G)TTG(A/T)AG-3′) was designed by Wang et al. (2006) and used to amplify the 3′-terminal fragment of the encoding gene (dv1). The 5′-terminal fragment of the gene was cloned by using the DNA Walking Speedup™ Premix Kit (Seegene, Korea). Three target specific primers (TSP) were designed according to the 3′-terminal fragment of the encoding gene (dv1) as follows: TSP1: 5-GACTTCTGGGGACTTG AGGA-3; TSP2: 5-CCAGAGTATCCCTTGAAACCA GAC-3; TSP3: 5-GAGTTGCGGTGGAAGCGAGA-3. Polymerase chain reaction (PCR) amplification was performed according to the user’s manual. All the PCR products were purified and sequenced by using an ABI PRISM 3730 automated sequencer (Perkin-Elmer, America) with four fluorescent dyes.
Analyses of nucleic acid and peptide sequences
Database searches and homologous analysis were performed using BlastX (http://www.ncbi.nlm.nih.gov/BLAST). Sequences were assembled using the SeqMan software (DNA Star software package). The promoter was predicted using the BDGP Neural network promoter prediction interface (http://www.fruitfly.org/seq_tools/promoter.html; Morton et al. 2003). Signal sequence prediction was performed using Signal P (http://www.cbs.dtu.dk/services/signalP/; Bendtsen et al. 2004). Protein molecular masses and isoelectric points were determined online, using ProtParam tools (http://us.expasy.org/tools/protparam.html). N-linked glycosylation sites were predicted by NetNGlyc (http://www.cbs.dtu.dk/services/NetNGlyc/).
Homology modeling
The sequence of Dv1 corresponding to the mature secreted protein, residues 123–407, was used in homology modeling. Homology modeling was done using the SWISS-PROT program (http://swissmodel.expasy.org//SWISS-MODEL.html; Guex and Peitsch 1997; Peitsch 1995; Schwede et al. 2003) and edited with the DeepView software (http://swissmodel.expasy.org/spdbv/). All residues but the last one was used in the modeling. Five proteins with known 3-D structures were used as templates in this process: four protease K proteins (2b6nA, 1p7vA, 1ic6A, 1p7wA) and one cold-adapted subtilisin-like serine protease (1sh7A; at the Protein Data Bank(http://www.rcsb.org/pdb).
Results
Protease production and purification
Four-liter culture filtrates were harvested by vacuum filtration and the protease was purified by chromatography. Purification factors and recoveries at each step were summarized in Table 1. The culture filtrate was concentrated by ultrafiltration. About 85.5% protease activity was recovered with a 1.7-fold purification (Table 1). The purified protease showed a single protein band on the 12% Coomassie Brilliant Blue R-250 stained gel. The molecular weight of the purified Dv1 was estimated to be 30 kDa by SDS-PAGE (Fig. 1).

SDS-PAGE electrophoresis gel. Lane 1, Purified Dv1. Lane M, protein marker
Effect of protease inhibitors and metal ions on the enzyme activity
The purified protease Dv1 was strongly inhibited by PMSF, indicating that it was a member of the serine protease family (Siezen and Leunissen 1997). Another serine protease inhibitor, Aprotinin showed a weak effect on Dv1 with 11.6% inhibition. The metal chelator EDTA inhibited the protease activity partially, likely because of the fact that EDTA can chelate Ca2+ ions. Ca2+ ions can confer thermo stability to proteases as has been demonstrated for the Bacillus Ak.1 protease (Smith et al. 1999). Aspartic protease inhibitors Pepstatin A and Leupeptin showed moderate effect on Dv1, with 14.2 and 21.5% inhibition, respectively. Metal ions Ca2+, Mg2+, Fe2+, and Cu2+ showed little effect on the activity of the purified protease. However, Zn2+ inhibited the enzyme activity (28%).
Effect of temperature and pH on enzyme activity
The optimum reaction temperature for Dv1 was 60.5°C, and the enzyme activity was stable when the temperature was below 40°C. However, the enzyme was inactivated at temperatures more than 70°C and with 20 min incubation. The protease showed the highest activity at pH 8.0. Between pH 6.0–11, the protease activity was stable. Bellow pH 7.0, the protease activity decreases with decreasing pH.
Hydrolysis of protein substrates and nematicidal activity
The purified protease showed a high hydrolytic activity against casein (100%); moderate hydrolysis of BSA (32%), gelatin (10%), and nematode cuticle (20%); and very little hydrolysis against collagen (3%).
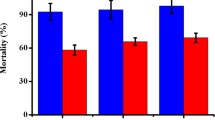
As shown in Table 2, both the crude extract and the purified protease showed obvious nematicidal activity against P. redivivus and C. elegans. Between the two host species, Dv1 was more active against C. elegans than P. redivivus. In the treatments, 50–100% tested nematodes were killed and degraded after being treated with either the crude extract or the purified enzyme for 12 h (Fig. 2). However, less than 20% of the tested nematodes were killed in the negative control sample. Furthermore, the crude extract showed a greater killing effect on nematodes than the purified enzyme, a result suggesting that other virulence factors in the crude extract may also play a role in the infection process.

Nematode treated with purified protease. a Control nematode. b–d Nematode (Panagrellus redivivus) treated with protease Dv1. Scale bar, 100 μm
Cloning of the serine protease Dv1
A 1.1 kb PCR product was amplified using the degenerate primers described in Materials and methods. The unknown 5′-terminal nucleotide sequence of dv1 was amplified using the DNA Walking Speedup™ Premix Kit, and a 1.3 kb PCR product was obtained. The full-length encoding gene of protease Dv1 was obtained by assembling sequences from the two PCR fragments with the DNA Star software package. The encoding gene of Dv1 consisted of one open-reading frame and without any intron.
A putative promoter from position-136 to position-185 upstream of the start codon (the A in the ATG being +1) was identified by online prediction, and a TATA box was identified at −176 bp upstream of the start codon. A similar TATA element was identified at −146 bp from the start codon of a serine protease in A. oligospora (Åhman et al. 1996). The most likely CAAT sequence and GC box in the 5′ untranslated region (UTR) of the dv1 gene were identified at −302 bp and at −316 bp upstream of the start codon, respectively. A diagram illustrating the features of the dv1 gene is shown in Fig. 3. The full nucleotide sequence of dv1 has been submitted to GenBank, under accession number DQ531603.

Diagram of Dv1 gene showing the presence of regulatory elements and ORF. The length of the sequence is 2449 bp. 5-UTR, 1–1225; Promoter, 1041–1090; TATA box, 1050–1055; CAAT box, 923–927; GC, 909–914; Signal peptide, 1226–1288; Proregion, 1289–1591; Mature peptide, 1592–2449; N terminus, 1592; Asp, 1712-1714; His, 1820-1800; Ser, 2279-2281
The encoding gene of Dv1 was typical of known fungal serine proteases (Fig. 3). It possesses a pre-pro-peptide structure, indicating that it is translated as a precursor polypeptide consisting of 407 amino acids with a calculated molecular mass of 42.0 kDa. It has a signal peptide (21 amino acids) consisting of the initial methionine, with the fourth amino acid an alkaline residue (Lys). It contains a core of eight hydrophobic residues (Gly-Ile-Ala-Thr-Phe-Ile-Thr-Phe-Ala-Gly-Leu-Ala) interrupted by two polar residues (Thr and Phe). There were three hydrophobic residues preceding the signal peptidase cleavage site (Ala-Leu-Ala). The pro-peptide cleavage site is before the N terminus of the secreted protein, and the final residue of the pro-peptide is an asparagine (N), at position-123 in Dv1. The first ten amino acids of the mature protease determined by our protein sequencer are AEQTDSTWGL, identical to the predicted N-terminal sequence of Dv1. The mature protein consists of 285 amino acids with a calculated molecular mass of 28.8 kDa and a theoretical pI of 8.47. The deduced amino-acid sequence of Dv1 had conserved motifs similar to those in subtilisin N and peptidase S8.
Modeling of Dv1
The three-dimensional structure of Dv1 was predicted using the Swiss-Prot method (Fig. 4a). All the sequences of the five templates used had more than 50% aa identities with Dv1. The molecule consisted of 18 beta-sheets and 6 alpha helices arranged in a single domain. The residues Asp41, His77, and Ser230 (in mature protease Dv1), located at the end of one beta-sheet and the beginning of two helices, respectively, make up the active site of the protease (Fig. 4a). Their locations in the molecular surface were shown in Fig. 4b.

a The ribbon modeling of the protease Dv1. The three conversed residues (serine 230, histidine 77, and asparagines 41) are at begins of two alpha helices and an end of a beta sheet, respectively. b Homology modeling of the molecular surface of Dv1 using five templates. The active site and substrate-binding region is indicated by a dashed black line. The colors (blue, green and yellow) on the surface relate to the accessibilities of residues to the aqueous environment, blue is least accessible and green to yellow are more accessible. Other colors’ meaning: red, serine 230; white, histidine 77; pink, asparagine 41
Comparison of Dv1 with other serine proteases from nematophagous fungi
The polypeptide sequence of Dv1 was aligned with other proteases from nematophagous fungi (Fig. 5). The deduced amino-acid sequence of Dv1 showed 62.5, 67.7, 66.4, 44.9, 42.1, 41.7, 44.5, and 44.1% identity, respectively, to Aoz1, Mlx, PII, VCP1, Ver112, PrA, Pr1, and pSP-3. The proregion cleavage positions of these enzymes were very conservative, and the first amino acid of these mature proteases was alanine. These proteases all had the conserved aspartic acid (Asp163)-histidine (His199)-serine (Ser352; in Dv1) catalytic triad. The two blocks of side chains that form the sides of the substrate-binding S1 pocket in subtilisin occur in regions of high sequence similarity and consisted of Ser258Leu259Gly260 and Ala284Ala285Gly286, respectively, in Dv1. Furthermore, the highly conserved Asn287 (in Dv1) was identified as important for the stabilization of the reaction intermediate during proteolysis by subtilisin (Kraut 1977).

Alignment of cuticle-degrading proteases amino acid sequences from D. varietes (Dv1), A. oligospora (PII and Aoz1), M. microscaphoides (Mlx), P. lilacinus (pSP-3), M. anisopliae (PrA), B. bassiana (Pr1), P. chlamydosporia (VCP1) and L. psalliotae (Ver112). The GenBank accession numbers of Dv1, PII, Aoz1, Mlx, pSP-3, PrA, Pr1, Ver112, and VCP1 are DQ531603, CAA63841, AAM93666, AAW21809, AAA91584, CAB64346, AAK70804, AAU01968, and CAD20578, respectively. Areas shaded in black are conserved regions (100% similarity), areas shaded in grey are high degree homology (more than 75% similarity), and unshaded areas are regions of variability between the proteases. Signal peptide sequences are marked on arrow, and triple asterisk indicates the N-terminal sequences of mature peptides. filled circle indicates the aspartic acid (Asp163)-histidine (His199)-serine (Ser352) (in Dv1) catalytic triad. Filled triangle indicates the N-linked glycosylation sites (Asn177) (in Dv1) and the underlined regions are the substrate-binding S1 pocket
As shown in Fig. 5, proteases PII, Aoz1, and Mlx from nematode-trapping fungi (those that form three-dimension nets) shared above 83% identity among each other. However, Dv1 only shared ∼65% aa identity to these three. Other three serine proteases VCP1, Ver112, and pSP-3 isolated from endoparasitic fungi (Pochonia chlamydosporia, Lecanicillium psalliotae and Paeciliomyces lilacinus) had more than 59% sequence identity to proteases PrA and Pr1 isolated from entomopathogenic fungi Metarhizium anisopliae and Beauveria bassiana, respectively. However, proteases (PII, Aoz1, Mlx, and Dv1) from nematode-trapping fungi shared a low sequence identity (42%) to those from endoparasitic and entomopathogenic fungi.
A phylogenetic tree (Fig. 6) was built based on the deduced peptide sequences from nematophagous and entomopathogenic fungi by the PHYLIP program package (Felsenstein 1991). Our analysis indicated that these serine proteases were clustered into two subclades. The four proteases (PII, Aoz1, Dv1, and Mlx) identified from nematode-trapping fungi formed one subclade, and the five proteases identified from endoparasitic fungi (Ver112, VCP1, and pSP-3) and entomopathogenic fungi (Pr1 and PrA) formed the second subclade.

Phylogenetic tree showing the relationship between Ac1 and other fungal subtilases. Phylogenetic analyses were performed with the PHYLIP program package (Felsenstein 1991). The data were subjected to Maximum Parsimony (MP) method of phylogenetic analysis, and the branch support of the MP tree was evaluated using bootstrap analysis with 1,000 replications. The GenBank accession numbers of proteases are described in Fig. 5. Aspergillus niger (accession number: AAA32703) was used as outgroup
Discussion
Serine proteases and other hydrolytic enzymes in nematophagous fungi are important virulence factors during infection against nematodes. Recently, several serine proteases have been identified from different nematophagous fungi (e.g., Bonants et al. 1995; Segers et al. 1994; Tunlid et al. 1994; Wang et al. 2006). These serine proteases shared some common properties: they have similar molecular weight (28–39 kDa); can be inhibited by PMSF; contains the conserved aspartic acid-histidine-serine catalytic triad and the substrate-binding site; and can degrade a broad range of substrates including casein, gelatin, BSA (bovine serum albumin), and nematode cuticle. In this report, a new pathogenic protease was isolated and characterized for the first time from the nematode-trapping fungus D. varietas. Our analysis identified that the N terminus amino-acid sequence of the mature protease Dv1 shared 90–100% sequence identity to PII, Aoz1, and Mlx, respectively. However, despite their high sequence similarities, Dv1 had biochemical properties different from these three proteases. On the contrary, the biochemical properties of Dv1 were more similar to VCP1, pSP-3, and Ver112, all isolated from parasitic fungi. For example, they all had a low molecular mass and a high pI. These results suggest that the genus Dactylellina may be a transitional group of organisms between nematode-trapping fungi and parasitic fungi.
Comparison of the dv1 gene with other fungal subtilase genes (Åhman et al. 1996; Joshi et al. 1995; St Leger et al. 1992) revealed regulatory elements upstream of the start codon (ATG; Fig. 3). A TATA box was identified at −176 bp from the start codon. Although its function has yet to be clearly determined in filamentous fungi, this box has been determined essential for the binding of RNA polymerase in higher eukaryotes during transcription (Morton et al. 2003). A putative promoter site (from 136 to 185) that includes the TATA box was identified and the transcription start site is within the 20–30 bp range of the TATA box that had been predicted for eukaryotic transcription start sites. The putative CAAT box, which plays a role in transcription initiation, was identified at position-302, and the GC box was identified in the 5′-UTR of the encoding gene, at position-316. Sequence alignments indicated that the 5′-UTR of PII and dv1 shared 49% nucleotide identity, lower than their identity in the translated region (69%).
The encoding genes of serine proteases from nematode-trapping fungi A. oligospora (PII and Aoz1) and M. microscaphoides (Mlx) contained one intron each (Åhman et al. 1996; Wang et al. 2006; Zhao et al. 2004), and the encoding genes of serine proteases from endoparasitic and entomopathogenic fungi P. chlamydosporia (VCP1), L. psalliotae (Ver112), M. anisopliae (PrA), and B. bassiana (Pr1) contained three introns each in their translated regions. However, the encoding gene of Dv1 lacked any intron in its translated region, different from other proteases from nematophagous and entomopathogenic fungi. To confirm its lack of intron, reverse transcription-PCR was done and identified that indeed no intron existed in the dv1 gene (data not shown). At present, the functions of these introns and the reason for the differences among these protease genes are still unknown.
The deduced primary sequence of the mature protease Dv1 contains one potential N-linked glycosylation site (Asn177). In both PII and Aoz1, there were two N-linked glycosylation sites (PII: Asn177 and Asn251; Aoz1: Asn178 and Asn252). However, the first N-linked glycosylation site of these two enzymes as well as Dv1 and Mlx (Asn175) were all very similar (Fig. 5). In contrast, Ver112, pSP-3, and VCP1 lacked any potential N-linked glycosylation site. This may have contributed to the larger molecular masses of proteases from nematode-trapping fungi than those from endoparasitic fungi.
The results in Table 2 showed that both the crude extract and the purified protease from D. varietas had obvious nematicidal effect on P. redivivus and C. elegans. These results suggest that the nematode-trapping fungus D. varietas may be used as a potential biocontrol agent against nematodes. Our analysis also indicated that the serine protease Dv1 identified from D. varietas showed several novel biochemical properties different from previous reported serine proteases. Therefore, our study enriched the information about infection-related extracellular serine proteases from nematophagous fungi. Moreover, the predicted three-dimensional structure of Dv1 provided baseline information for further studies on the relationships between structure and function.
References
Åhman J, Ek B, Rask L, Tunlid A (1996) Sequence analysis and regulation of a gene encoding a cuticle-degrading serine protease from the nematophagous fungus Arthrobotrys oligospora. Microbiology 142:1605–1616
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: signalP 3.0. J Mol Biol 340:783–795
Bonants PJ, Fitters PF, Thijs H, den Belder E, Waalwijk C, Henfling JW (1995) A basic serine protease from Paecilomyces lilacinus with biological activity against Meloidogyne hapla eggs. Microbiology 141:775–784
Bradford MM (1976) A rapid and sensitive for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Cox GN, Kusch M, Edgar RS (1981) Cuticle of Caenorhabditis elegans: its isolation and partial characterization. J Cell Biol 90:7–17
Felsenstein J (1991) PHYLIP: phylogeny inference package, version 3.5. University of Washington, Seattle, WA
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis 18:2714–2723
Huang XW, Zhao NH, Zhang KQ (2004) Extracellular enzymes serving as virulence factors in nematophagous fungi involved in infection of the host. Res Microbiol 155:811–816
Jansson HB, Nordbring-Hertz B (1988) Poinar GO Jr, Jansson H-B (eds) Infection mechanisms in the fungus-nematode system in diseases of nematodes, vol. II. CRC, Boca Raton, pp 59–72
Joshi L, St Leger RJ, Bidochka MJ (1995) Cloning of a cuticle-degrading protease from the entomopathogenic fungus, Beauveria bassiana. FEMS Microbiol Lett 125:211–217
Kraut J (1977) Serine proteases structure and mechanism of catalysis. Annu Rev Biochem 46:331–358
Laemmli EK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Li Y, Jeewon R, Hyde KD, Mo MH and Zhang KQ (2006) Two new species of nematode-trapping fungi: relationships inferred from morphology, rDNA and protein gene sequence analyses. Mycol Res 110:790–800
Lopez-Llorca LV (1990) Purification and properties of extracellular protease produced by the nematophagous fungus Verticillium suchlasporium. Can J Microbiol 36:530–537
Maizels RM, Blaxter ML, Selkirk ME (1993) Forms and functions of nematode surfaces. Exp Parasitol 77:380–384
Morton CO, Hirsch PR, Peberdy JP, Kerry BR (2003) Cloning of and genetic variation in protease VCP1 from the nematophagous fungus Pochonia chlamydosporia. Mycol Res 107:38–46
Peitsch MC (1995) Protein modeling by e-mail. Bio/Technology 13:658–660
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Segers R, Butt TM, Kerry BR, Peberdy JF (1994) The nematophagous fungus Verticillium chlamydosporium produces a chymoelastase-like protease which hydrolyses host nematode proteins in situ. Microbiology 140:2715–2723
Siddiqui ZA, Mahmood I (1996) Biological control of plant parasitic nematodes by fungi: a review. Bioresour Technol 58:229–239
Siezen RJ, Leunissen JAM (1997) Subtilases: the superfamily of subtilisin like serine protease. Protein Sci 6:501–523
Smith CA, Toogood HS, Baker HM, Daniel RM, Baker EN (1999) Calcium-mediated thermostability in the subtilisin superfamily: the crystal structure of Bacillus Ak.1 protease at 1.8 Å resolution. J Mol Biol 294:1027–1040
St Leger RJ, Frank DC, Roberts DW, Staples RC (1992) Molecular cloning and regulatory analysis of the cuticle-degrading protease structural gene from the entomopathogenic fungus Metarhizium anisopliae. Eur J Biochem 204:991–1001
Subramanian CV (1963) Dactylella, Monacrosporium and Dactylina. J Indian Bot Soc 42:291–300
Tunlid A, Jansson S (1991) Proteases and their involvement in the infection and immobilization of nematodes by the nematophagous fungus Arthrobotrys oligospora. Appl Environ Microbiol 57:2868–2872
Tunlid A, Rosen S, EK B, Rask L (1994) Purification and characterization of an extracellular serine protease from the nematode-trapping fungus Arthrobotrys oligospora. Microbiology 140:1687–1695
Wang M, Yang JK, Zhang KQ (2006) Characterization of an extracellular protease and its cDNA from the nematode-trapping fungus Monacrosporium microscaphoides. Can J Microbiol 52:130–139
Yang JK, Huang XW, Tian BY, Wang M, Niu QH, Zhang KQ (2005a) Isolation and characterization of a serine protease from the nematophagous fungus Lecanicillium psalliotae, displaying nematicidal activity. Biotechnol Lett 27:1123–1128
Yang JK, Huang XW, Tian BY, Sun H, Duan JX, Wu WP, Zhang KQ (2005b) Characterization of an extracellular serine protease gene from the nematophagous fungus Lecanicillium psalliotae. Biotechnol Lett 27:1329–1334
Zhang D, Yang Y, Castlebury LA, Cerniglia CE (1996) A method for the large scale isolation of high transformation efficiency fungal genomic DNA. FEMS Microbiol Lett 145:216–265
Zhao ML, Mo MH, Zhang KQ (2004) Characterization of a neutral serine protease and its full-length cDNA from the nematode-traping fungus Arthrobotrys oligospora. Mycologia 96:16–22
Acknowledgement
We thank Drs. Chenggang Zhou, Xiaowei Huang and Ms Wei Zhou for their help and advice in our studies and in preparing this manuscript. This work was funded by projects from the National Natural Science Foundation of China (approved nos. 30630003 and 30660107), by the Department of Science and Technology of Yunnan Province (approval Nos. 2004C0001Z and 2005NG05) and by Yunnan University (grant no. 2005Q008B).
Author information
Authors and Affiliations
Corresponding author
Additional information
Jinkui Yang and Lianming Liang contributed equally to this work.
Rights and permissions
About this article
Cite this article
Yang, J., Liang, L., Zhang, Y. et al. Purification and cloning of a novel serine protease from the nematode-trapping fungus Dactylellina varietas and its potential roles in infection against nematodes. Appl Microbiol Biotechnol 75, 557–565 (2007). https://doi.org/10.1007/s00253-007-0839-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-007-0839-6