
Abstract
Twenty-two Bacillus cereus strains were screened for phospholipase C (PLC, EC 3.1.4.3) activity using p-nitrophenyl phosphorylcholine as a substrate. Two strains (B. cereus SBUG 318 and SBUG 516) showed high activity at elevated temperatures (>70°C) at acidic pH (pH 3.5–6) and were selected for cloning and functional expression using Bacillus subtilis. The genes were amplified from B. cereus DNA using primers based on a known PLC sequence and cloned into the expression vector pMSE3 followed by transformation into B. subtilis WB800. On the amino acid level, one protein (PLC318) was identical to a PLC described from B. cereus, whereas PLC516 contained an amino acid substitution (E173D). PLC production using the recombinant strains was performed by an acetoin-controlled expression system. For PLC516, 13.7 U g−1 wet cell weight was determined in the culture supernatant after 30 h cultivation time. Three purification steps resulted in pure PLC516 with a specific activity of 13,190 U mg−1 protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phospholipase C (PLC) cleaves phospholipids stereospecifically into sn1,2-diacylglycerides and phosphate monoesters (El-Sayed and Roberts 1985). Some PLCs are more substrate-specific than others. For example, the inositol-specific PLC (E.C.3.1.4.10) cleaves only phosphatidylinositol-4,5-bisphosphate into sn1,2-diacylglycerides and inositol-(1,4,5)-triphosphate, the latter being an important second messenger in calcium metabolism (Tan and Roberts 1998). In contrast, the phosphatidylcholine-preferring phospholipase C (PC-PLC; E.C.3.1.4.3) prefers phosphatidylcholine but can accept other phospholipids as well (Martin and Hergenrother 1998). Due to its high structural and catalytic similarities to mammalian PLCs, the enzyme from Bacillus cereus was also the subject of medical research (Garcia de Herreros et al. 1991; Mulcahy et al. 1985). Moreover, this enzyme was used in basic research in the areas of lipid metabolism, chemistry of lipids, blood coagulation and of eukaryotic cell membranes (Kamberov and Ivanov 1990). This might explain why PC-PLCs are the most thoroughly investigated bacterial enzymes of their class. The physiological function of the PC-PLC is not known, but the enzyme was reported to be part of a phosphate retrieval system (Guddal et al. 1989).
The PC-PLC from B. cereus is an extracellular, monomeric tri-zinc protein (Little and Otnaess 1975) without an intracellular pool (Toora and Singh 1990). It is synthesized as a 283-amino acid long pre-proenzyme from which the 24-aa long presequence and the 14-aa long prosequence are cleaved first. The mature enzyme has a length of 245 aa and a calculated molecular weight of 28,520 Da, including the three zinc ions, which is closely in line with the data obtained on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; ∼27 kDa). The PC-PLC gene has a length of 852 nucleotides and it is located on the bacterial chromosome (Johansen et al. 1988). Expression of the PC-PLC gene in Escherichia coli lead to a high amount of inclusion bodies (Tan et al. 1997). In this case, a cumbersome refolding, purification, and activation protocol is necessary to get active enzyme. The overexpression of the PC-PLC gene in Pichia pastoris works much better and can lead to large amounts of active enzyme in the supernatant (Seo and Rhee 2004), but cultivations are relatively long and the system used is due to licensing fees.
The PC-PLC from B. cereus shows activity in aqueous systems (Hergenrother et al. 1995), in two-phase systems (Haftendorn and Ulbrich-Hofmann 2002), and in organic solvents (Anthonsen et al. 1999). Thus, it can be used in a wide range of reaction systems with the synthesis of optically pure sn1,2-diglycerides as example, as these compounds can be used as phospholipid precursors for the pharmaceutical industry (Anthonsen et al. 1999).
In this paper, we describe the screening and isolation of two PLC from B. cereus and the cloning and expression of the corresponding genes. As B. cereus strains can be pathogenic (Ghelardi et al. 2002; Notermans and Batt 1998), quasi-homologous expression in the closely related Bacillus subtilis was performed.
Materials and methods
All chemicals and kits were obtained at the highest purity available from common commercial suppliers. The B. cereus strains were from the strain collection of the Institute of Microbiology at Greifswald University (SBUG). p-Nitrophenylphosphorylcholine (NPPC) was synthesized as described before (Durban and Bornscheuer 2003). For the overexpression of the PLC gene from B. cereus, an acetoin-regulated B. subtilis expression system was used (Silbersack et al. 2006). The PLC genes were cloned and overexpressed in B. subtilis strain WB800 (nprE, aprE, epr, bpr, mpr::ble, nprB::bsr, vpr, wprA::hyg), which is deficient in eight extracellular proteases (Wu et al. 2002).
p-Nitrophenylphosphorylcholine assay
Ten microliters of a 100-mM solution of NPPC in borax–HCl buffer (100 mM, pH 5–9) and 90 μl of an enzyme solution (lyophilized PLC redissolved in borax–HCl buffer,100 mM, pH 5–9) were mixed in a well of a microtiter plate. The p-nitrophenol released was then quantified by measurement of its absorbance at 410 nm for 30 min at 37°C in 30-s steps in a microtiter plate reader (FLUOstar galaxy, BMG, Offenburg, Germany; see Kurioka and Matsuda 1976). Calibration curves were created with varying concentrations of p-nitrophenol and at different pH values. At temperatures higher than 45°C, 100 μl of a 100-mM solution of NPPC in borax–HCl buffer (100 mM, pH 5–9) and 900 μl of an enzyme solution (lyophilized PLC redissolved in borax–HCl buffer, 100 mM, pH 5–9) were mixed in a reaction tube and incubated at different temperatures in a thermoshaker (Thermomixer comfort, Eppendorf, Hamburg, Germany). Every 4 min a sample of 100 μl was taken and the p-nitrophenol released was then quantified as described above.
Cultivation of B. cereus strains
The 22 B. cereus strains were cultivated in 2-l shake flasks containing 500 ml Luria–Bertani (LB) medium. The LB medium was inoculated with a 5-ml overnight culture of each B. cereus strain and cultivated for 10 h in a shaking incubator (Unitron, Infors, Basel, Switzerland, 37°C, 150 rpm). The cells were separated from the medium by centrifugation (Multifuge 3 S-R, Kendro, Hanau, Germany; 15 min, 4°C, 4,570×g), and the proteins in the supernatant (450 ml) were concentrated with centricons (pore size 30 kDa, Millipore, Billerica, MA, USA). The concentrated enzyme solution was freeze-dried. The lyophilized crude protein extract containing PLC was used for further characterizations. The activity of these PLCs was determined with the NPPC assay as described above.
Cloning of the PLC genes
The DNA of B. cereus SBUG 516 was isolated with a commercially available DNA isolation kit. Polymerase chain reaction (PCR) primers were designed based on the known sequence (accession code: X12854) of a PLC from B. cereus (Johansen et al. 1988). At the 3′-end, the TOPO recognition sequence and a XbaI site and at the 5′-end, an overhang for the subsequent fusion PCR was created [Fus-acoA-amyE-mPLC (5′-GGC TGC GAG TGC TTG GTC TGC TGA AG-3′) and PLC-RW-Xba (5′-CCG TCT AGA TTA ACG ATC TCC GTA CG-3′)]. The gene (mPLC) was amplified using a standard PCR protocol. The acoA-promoter/amyE-signal peptide fusion sequence (acoAamyE) was amplified using primers, which make it possible to create an overhang for the following fusion PCR at the 3′- end and an XbaI site at the 5′- end [Xba-Topo (5′-CAC CTC TAG ATC AGT CAA ACG ATG-3′) and Fus_mPLC_aco_amy_RW (5′-CTT CAG CAG ACC AAG CAC TCG CAG CC-3′)]. The two sequences were fused and cloned into a TOPO vector. After transformation of the plasmid into E. coli XL 21 blue, the plasmid was purified and digested with XbaI. The 1,100-kb fragment containing the fused acoAamyEmPLC sequence was ligated into a XbaI-cut pMSE3 (Silbersack et al. 2006). The resulting plasmid pMSE3-516 (Fig. 1) was transformed into E. coli DH5α and sequenced. The plasmid was purified and transformed into B. subtilis WB800 (Wu et al. 2002). In a similar manner, the gene encoding for PLC318 was identified and cloned.

Plasmid map of the vector pMSE3-516 used for overexpression of the PLC gene from B. cereus 516 (acoA promoter of the acoABCL operon from B. subtilis, amyE alpha-amylase signal sequence from B. subtilis, PLC gene for the PLC from B. cereus 516, ColE1 origin of replication of E. coli, aphA3 kanamycin resistance from Campylobacter coli, res beta resolvase gene and repE replication initiation protein from pAMβ1, a plasmid from Enterococcus faecalis)
Cultivation of B. subtilis WB800 and expression of the PC-PLC
Cultivations were performed in SB medium (32 g l−1 tryptone, 20 g l−1 yeast extract, 5 g l−1 NaCl, 5 ml l−1 1 N NaOH). After sterilization, 900 ml of this solution was mixed with 100 ml of a sterile salt solution (120 g l−1 Na2HPO4, 60 g l−1 KH2PO4, 20 g l−1 NH4Cl, 60 mg l−1 CaCl2). For cultivation of B. subtilis WB800 bearing the plasmids pMSE3-516 or pMSE3-318, a 50-ml preculture was prepared from a 5-ml overnight culture. This preculture was cultivated at 37°C and 150 rpm until an OD600 = 0.85 was reached. This preculture was used to inoculate 4-L SB medium in a fermenter (New Brunswick Scientific, Edison, NJ, USA). Cultivation was then performed at 37°C and pH 6.5. Agitation and air flow were controlled by the oxygen content in the medium, which was fixed to 20% dO2. The expression of the PLC was induced at an OD600 = 3 by adding acetoin (final concentration 0.5 % v/v). After 30 h, cells were separated from the media by centrifugation (Multifuge 3 S-R, Kendro, Hanau, Germany; 15 min, 4°C, 4,570×g), and the proteins in the supernatant (4 l) were salted out with 90% ammonium sulfate. The precipitated proteins were redissolved in borax–HCl buffer (100 mM, pH 7.5) and concentrated with centricons (Centricon Plus-20, 30 kDa, Millipore, Bedford, MA, USA). The PLC was located in the retentate, while the flow-through showed no PLC-activity. The retentate was purified by gel filtration (Sephacryl S-200 High Resolution) using an Äktaprime device (Amersham Biosciences; flow rate 2 ml min−1, fraction size 2 ml, borax–HCl buffer, 100 mM, pH 7.5). Active fractions containing the PLC were concentrated with centricons, again to reduce the volume.
To compare the wild-type enzyme to the recombinant one, B. cereus SBUG 516 and SBUG 318 wild-type strains were cultivated under the conditions described above until the highest PLC activity in the supernatant was reached. The enzymes were purified as described above. All PLC activities were determined with the NPPC assay as described above.
Gel electrophoresis
Sodium dodecyl sulfate (SDS) gels were prepared as described (Sambrook et al. 1989).
Results
Identification and characterization of PLCs from different B. cereus strains
Twenty-two B. cereus strains were cultivated in LB medium, and 12 strains produced high PLC activity in the culture supernatant as estimated from the activity assay using p-nitrophenylphosphorylcholine (NPPC) as substrate. Further characterization revealed that all enzymes show highest activity at an acidic pH between 3.5 and 6, and most of them were found active in a temperature range between 20 and 60°C (data not shown). However, two enzyme preparations obtained from B. cereus SBUG 318 and SBUG 516 exhibited activity >70°C and were chosen for subsequent cloning and expression.
Cloning of the PLC genes
After DNA isolation from the corresponding B. cereus strains, amplification of the genes encoding the PLCs 318 and 516 was straightforward due to the availability of a known sequence (Tan et al. 1997). Sequence analysis revealed that the PLC-encoding gene from B. cereus SBUG 516 differed from the published sequence in 13 nucleotides, which translated into one amino acid substitution (E173D) in the mature protein. The PLC318 gene contained six nucleotide substitutions, but the protein had an identical sequence to the published PC-PLC. Therefore, the properties of PLC318 were not analyzed in detail, and further experiments concentrated on PLC516.
Expression of the PLCs in B. subtilis and purification of the enzyme from strain SBUG 516
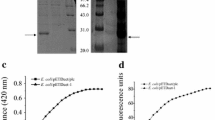
The quasi-homologous expression of both recombinant PLCs (from B. cereus SBUG 318 and SBUG 516) in B. subtilis WB800 using plasmid pMSE3 (Fig. 1) turned out to be an easy way to produce high amounts of active enzyme. As exemplified for PLC318, the PLC activity in the medium constantly increased after induction of the acoA-directed expression system with acetoin up to 200 U ml−1 medium after 30 h (Fig. 2), while with the best wild-type strain a maximum activity of 35 U ml−1 medium was reached (Fig. 2). Furthermore, in contrast to the enzyme production in the wild-type strain, no degradation of the produced PLC after a prolonged cultivation time was observed. No significant differences between expression of PLC318 and PLC516 were observed. The crude culture supernatant from a PLC516 cultivation was then subjected to a three-step purification method resulting in highly purified PLC (Table 1, Fig. 3) with a specific activity of 13 190 U mg−1 protein. The purified protein was also subjected to matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) analysis after tryptic digestion, which confirmed its identity as a phospholipase C (data not shown).

Comparison of growth curves (triangle), PLC activity (square), and wet cell weight (diamond) of cultivations of B. subtilis WB 800 containing plasmid pMSE3-516 (black symbols) and the B. cereus wild-type strain 318 (white symbols)

SDS-PAGE gel of different fractions of the gel filtration step with Sephacryl S-200 High Resolution. Lines 1 to 7 shows the successive most active fractions, while lines 8 and 9 show the enzyme solution after concentration with centricons (1:10 dilution of the sample). The gel was stained using Coomassie Brilliant Blue R250. The band of line 4 was cut off and used for MALDI-TOF analysis
Characterization of the recombinant PLC516
Subsequently, the native PLC from the wild-type strain cultivation and the recombinant enzyme were compared. Only slight differences in the temperature profiles of the enzymes from B. cereus 516 were observed (Fig. 4). The recombinant PLC had its maximum activity at 60°C, which is 5°C lower than for the wild-type enzyme, which also appeared to be slightly more stable at higher temperatures. Similarly, the pH profiles of the different preparations of PLC516 were identical (Fig. 5).

Temperature profiles of PLC516. Wild-type (circle); recombinant, crude extract (square); recombinant, after gel filtration (diamond)

pH profiles of PLC516. Wild-type (circle); recombinant, crude extract (square); recombinant, after gel filtration (diamond)
Discussion
Different methods for a recombinant expression of a PLC from B. cereus have been described. As already pointed out in the “Introduction,” expression in E. coli lead to the formation of large amounts of inactive enzyme as inclusion bodies, which can only be converted into active protein by a lengthy protocol with low yields (Tan et al. 1997). Another possibility was the expression in P. pastoris (Seo and Rhee 2004), which yielded active crude protein with a specific activity of 678 U mg−1 and 268–365 U ml−1 culture volume. We could show in this work that PLC can also be functionally produced and secreted in B. subtilis using a novel expression system (Silbersack et al. 2006). This expression system, which can be easily modulated by the acetoin concentration in the medium, was successfully applied to two PLCs identified from the screening of B. cereus wild-type strains. Moreover, high productivities (approx. 200 U ml−1 culture volume) are possible after reasonable cultivation times (∼30 h). In addition, a simple purification protocol yields highly pure PLC. It should be emphasized that the GRAS status of the B. subtilis strain used also allows for the application of the recombinant PLCs in food production.
References
Anthonsen T, D’Arrigo P, Pedrocchi-Fantoni G, Secundo F, Servi S, Sundby E (1999) Phospholipids hydrolysis in organic solvents catalysed by immobilised phospholipase C. J Mol Catal B Enzym 6:125–132
Durban M, Bornscheuer UT (2003) An assay system for the detection of phospholipase C activity. Eur J Lipid Sci Technol 105:633–637
El-Sayed MY, Roberts MF (1985) Charged detergents enhance the activity of phospholipase C (Bacillus cereus) towards micellar short-chain phosphatidylcholine. Biochim Biophys Acta 831:133–141
Garcia de Herreros A, Dominguez I, Diaz-Meco MT, Graziani G, Cornet ME, Guddal PH, Johansen T, Moscat J (1991) Requirement of phospholipase C-catalyzed hydrolysis of phosphatidylcholine for maturation of Xenopus laevis oocytes in response to insulin and ras p21. J Biol Chem 266:6825–6829
Ghelardi E, Celandroni F, Salvetti S, Barsotti C, Baggiani A, Senesi S (2002) Identification and characterization of toxigenic Bacillus cereus isolates responsible for two food-poisoning outbreaks. FEMS Microbiol Lett 208:129–134
Guddal PH, Johansen T, Schulstad K, Little C (1989) Apparent phosphate retrieval system in Bacillus cereus. J Bacteriol 171:5702–5706
Haftendorn R, Ulbrich-Hofmann R (2002) Activity of phospholipase C in two-phase systems. Anal Biochem 306:144–147
Hergenrother PJ, Spaller MR, Haas MK, Martin SF (1995) Chromogenic assay for phospholipase C from Bacillus cereus. Anal Biochem 229:313–316
Johansen T, Holm T, Guddal PH, Sletten K, Haugli FB, Little C (1988) Cloning and sequencing of the gene encoding the phosphatidylcholine-preferring phospholipase C of Bacillus cereus. Gene 65:293–304
Kamberov E, Ivanov A (1990) Purification of phospholipase-C from Bacillus cereus by affinity chromatography on 2-(4-aminophenylsulphonyl)ethyl-cellulose. J Chromatogr 525:307–318
Kurioka S, Matsuda M (1976) Phospholipase C assay using p-nitrophenylphosphoryl-choline together with sorbitol and its application to studying the metal and detergent requirement of the enzyme. Anal Biochem 75:281–289
Little C, Otnaess A-B (1975) The metal ion dependence of phospholipase C from Bacillus cereus. Biochim Biophys Acta 391:326–333
Martin SF, Hergenrother PJ (1998) General base catalysis by the phosphatidylcholine-preferring phospholipase C from Bacillus cereus: the role of Glu4 and Asp55. Biochemistry 37:5755–5760
Mulcahy LS, Smith MR, Stacey DW (1985) Requirement for ras proto-oncogene function during serum-stimulated growth of NIH 3T3 cells. Nature 313:241–243
Notermans S, Batt CA (1998) A risk assessment approach for food-borne Bacillus cereus and its toxins. Symp Ser Soc Appl Microbiol 84:51–61
Sambrook J, Fritsch EF, Manniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press
Seo K-H, Rhee JI (2004) High-level expression of recombinant phospholipase C from Bacillus cereus in Pichia pastoris and its characterization. Biotechnol Lett 26:1475–1479
Silbersack J, Jürgen B, Hecker M, Schneidinger B, Schmuck R, Schweder T (2006) An acetoin-regulated expression system of Bacillus subtilis. Appl Microbiol Biotechnol (in press). DOI https://doi.org/10.1007/s00253-006-0549-5
Tan CA, Roberts MF (1998) Engineering of the nonspecific phospholipase C from Bacillus cereus: replacement of glutamic acid-4 by alanine results in loss of interfacial catalysis and enhanced phosphomonoesterase activity. Biochemistry 37:4275–4279
Tan CA, Hehir MJ, Roberts MF (1997) Cloning, overexpression, refolding, and purification of the nonspecific phospholipase C from Bacillus cereus. Protein Expr Purif 10:365–372
Toora S, Singh G (1990) Factors affecting production and activity of phospholipase C by Yersinia enterocolitica. J Appl Bacteriol 69:113–118
Wu SC, Yeung JC, Duan Y, Ye R, Szarka SJ, Habibi HR, Wong SL (2002) Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl Environ Microbiol 68:3261–3269
Acknowledgements
We are grateful to Südchemie (München, Germany) for financial support and to Dr. Dirk Albrecht from the Division of Microbial Physiology and Molecular Biology of the Ernst-Moritz-Arndt-University Greifswald for the MALDI-TOF mass spectrometry analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Durban, M.A., Silbersack, J., Schweder, T. et al. High level expression of a recombinant phospholipase C from Bacillus cereus in Bacillus subtilis . Appl Microbiol Biotechnol 74, 634–639 (2007). https://doi.org/10.1007/s00253-006-0712-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0712-z