
Abstract
The Bacillus subtilis lipoprotein PrsA enhances the yield of several homologous and heterologous exported proteins in B. subtilis by being involved in the posttranslocational stage of the secretion process. In this work, we have studied the effect of B. subtilis PrsA on the secretion of Bacillus amyloliquefaciens α-amylase (AmyQ), a target protein for PrsA, and Bacillus licheniformis penicillinase (PenP) a nontarget protein for PrsA, in Lactococcus lactis. Two compatible plasmids were constructed and introduced into L. lactis strain NZ9000: one high copy plasmid, expressing the AmyQ gene (amyQ) or the PenP gene (penP), and one low copy plasmid, expressing the PrsA encoding gene (prsA). When amyQ and prsA were simultaneously expressed under the nisin-inducible promoter PnisA, Western blotting experiments revealed a 15- to 20-fold increase in the total yield of AmyQ and a sixfold increase in secreted AmyQ activity, compared to a control strain lacking prsA. When expressed under the same induction conditions, PrsA had no effect on the secretion or total yield of PenP. These results show that the secretion yield of some heterologous proteins can be significantly increased in L. lactis when coproduced with the B. subtilis PrsA protein.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lactococcus lactis is the most extensively studied lactic acid bacteria (LAB). It is classified as a GRAS (generally regarded as safe) bacterium and is widely used in the food industry. Recently, production of heterologous proteins in LAB, for use as enzymes, therapeutics, and vaccines, has become a research field of interest and, hence, optimization of gene expression and protein secretion efficiencies in L. lactis has become important. Many systems for protein production and secretion were designed during the last decade. However, even if some production systems for high-level production and secretion of heterologous proteins were reported (Bermudez-Humaran et al. 2003; Lindholm et al. 2004; Novotny et al. 2005; Savijoki et al. 1997), the acquired production yields of heterologous proteins have mostly been rather low in L. lactis. Therefore, it is important to further explore possibilities to improve protein production and secretion in this important protein production host.
PrsA of Bacillus subtilis is a 33-kDa lipoprotein that is anchored to the outer surface of the cytoplasmic membrane through a thiol-linked diacylglycerol in the N-terminal cysteine residue (Kontinen and Sarvas 1993; Kontinen et al. 1991; Leskela et al. 1999). In B. subtilis, PrsA exercises a chaperon-like function and the increase in PrsA production has been shown to decrease the degradation of some exported proteins resulting in increased yields of secreted protein in the culture medium (Sarvas et al. 2004).
In this work we have studied the effect of B. subtilis PrsA on the secretion of heterologous proteins in L. lactis. For this, two different heterologous proteins were chosen, Bacillus amyloliquefaciens α-amylase (AmyQ) as a target protein for PrsA and Bacillus licheniformis penicillinase (PenP) as a nontarget protein for PrsA (Kontinen and Sarvas 1988; Vitikainen et al. 2005). Both proteins were tested in the same production system with and without PrsA. Our experiments showed that coinduction of prsA and amyQ with an optimal nisin concentration significantly increased the total yield of AmyQ compared to the control strain lacking prsA. In agreement with studies on B. subtilis, PrsA did not have any effect on PenP secretion in L. lactis.
Materials and methods
Bacteria, plasmids, and growth conditions
Bacterial strains and plasmids are listed in Table 1. Escherichia coli and B. subtilis were grown in Luria–Bertani medium (Difco) at 37 °C and L. lactis in M17 medium (Difco) supplemented with 0.6% glucose (GM17) at 30 °C. Chloramphenicol (5–10 μg/ml), erythromycin (5 μg/ml), and kanamycin (5 μg/ml) were added as indicated in Table 1. For pulse chase experiments, L. lactis was grown in chemically defined medium (CDM) (Otto et al. 1983; Poolman and Konings 1988) depleted of methionine and supplemented with 1% glucose. Growth rate as a function of time was determined by optical density measurement at 600 nm (OD600).
DNA methods and plasmid constructions
Primers used for DNA amplification are listed in Table 2. A DNA fragment containing the promoter sequence (PslpA) and export signal sequence (SSslpA) of the surface layer protein gene slpA was polymerase chain reaction (PCR)-amplified from the chromosomal DNA of Lactobacillus brevis ATCC 8287 using primers P1 and P2. The PCR fragment was inserted into the EcoRI and BamHI cut plasmid pKTH2121 (Savijoki et al. 1997) upstream of the transcription terminator sequence of slpA (tslpA) present in the plasmid. The fragment SSslpA-(XhoI-BamHI)-tslpA was then PCR amplified from the resulting plasmid (pKTH2139) with primers P3 and P4 and cloned into the NcoI-HindIII site of pNZ8032 (de Ruyter et al. 1996b) to construct pKTH5036, which was used to express the genes coding for B. amyloliquefaciens AmyQ and B. licheniformis PenP under the NisA promoter (PnisA) in L. lactis. The AmyQ gene (amyQ, GenBank accession no. J01542; Takkinen et al. 1983) was PCR-amplified from the plasmid pKTH10 (Palva 1982) using primers P5 and P6 (Table 2) and the PenP gene (penP, GenBank accession no. V00093; Neugebauer et al. 1981) was PCR-amplified from the chromosomal DNA of B. licheniformis with primers P7 and P8. The secretion plasmids pKTH5041 (amyQ) and pKTH5054 (penP) were constructed by cloning the reporter genes into the XhoI site of pKTH5036. E. coli was used as an intermediate cloning host for the reporter genes. For pKTH5066 with the prsA gene under control of PnisA, an intermediate plasmid with the PnisA-prsA fusion was constructed. The prsA gene was PCR-amplified from the chromosomal DNA of B. subtilis with the primer pair P9 and P10 and cloned to the NcoI site of pNZ8037 (de Ruyter et al. 1996b) to construct pKTH2167. The PnisA-prsA fusion segment was then amplified with PCR from this plasmid using primers P11 and P12 and cloned into the BamHI site of pIL277 (Simon and Chopin 1988) resulting in pKTH5066. All PCR amplifications were performed with Dynazyme II polymerase (Finnzymes). The transformation of L. lactis strains was carried out as described by Holo and Nes (1995). All the plasmid constructs were proven to be correct by nucleotide sequence analysis using an Applied Biosystems (ABI) Prism 310 genetic analyzer with a Big Dye terminator version 3.1 cycle sequencing kit (Applied Biosystems, Foster City, USA).
Nisin induction and preparation of cellular and supernatant fractions
The expression of amyQ, penP, and prsA in L. lactis induced by various nisin concentrations was tested. Samples were withdrawn from nisin-induced cell cultures at different time points and the OD600 values of the cultures were determined. Cells were separated by centrifugation (2,000×g, 10 min at 4 °C) and supernatant fractions were used directly for enzyme activity assays and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The cell fractions were washed with PBS and disrupted with glass beads by cell mill homogenization (Bühler Vibrogen Cell Mill VL4). The cell homogenate was resuspended in 10 mM N-(2-hydroxyethyl)-piperazine-N′-2-ethanesulfonic acid (HEPES buffer), pH 7, and was used for activity assays and immunoblotting.
Enzyme activity assays
Determination of AmyQ activity in supernatant and cell fractions was carried out using Phadebas® Amylase Test tablets (Pharmacia and Upjohn Diagnostics AB) and AmyQ concentration was calculated from the activity of a purified AmyQ standard (Sigma A-6380). PenP assay was performed using Nitrocefin (Oxoid) according to the manufacturer’s instructions. PenP activity units were calculated using the formula \( U = \Delta \;{A_{{480}} } \mathord{\left/ {\vphantom {{A_{{480}} } {t_{{\min }} \times 26.3}}} \right. \kern-\nulldelimiterspace} {t_{{\min }} \times 26.3} \) (Simons et al. 1978).
SDS-PAGE and immunoblotting
For quantitative analysis of AmyQ and PrsA, SDS-PAGE sample buffer was added to an equal volume of nisin-induced cell and supernatant fractions before boiling (5 min) and separation of proteins by SDS-PAGE electrophoresis. Proteins were transferred to a nitrocellulose Trans-Blot transfer membrane (BioRad) and visualized by rabbit antisera against AmyQ or PrsA, horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin IgG (BioRad), and SuperSignal® West Dura Extended Duration Substrate (Pierce). The chemiluminiscence generated was detected using phosphor screen technology on a Molecular Imager G-525 system (BioRad) and the protein bands were quantified using the Quantity One software (BioRad). Rabbit antisera against AmyQ and PrsA were provided by Dr. Ilkka Palva and Dr. Vesa Kontinen, respectively. The amount of PrsA production in L. lactis was quantified in Western blots with the help of a purified PrsA standard, provided by Dr. Vesa Kontinen. The proteins of the [35S]methionine (MET)-labeled and immunoprecipitated samples were separated on a 4–12% NuPAGE® Bis–Tris precast gel (Novex) according to the manufacturer’s instructions. The gels were dried, photographed on a FluorS MultiImager apparatus (BioRad) and the protein bands were quantified as described above.
Preparation of protoplasts
Expression of amyQ and prsA in NZ9000(pKTH5041+pKTH5066) and amyQ in NZ9000(pKTH5041+pIL277) was induced with 1 or 10 ng/ml nisin at OD600 0.3–0.4. After 5 h of induction at 30 °C, the cells were harvested by centrifugation (5,000×g, 10 min) and washed in 50 mM HEPES buffer (pH 7). One milliliter of washed cells was withdrawn before resuspension of cells in protoplast buffer (20 mM HEPES, pH 7; 40 mM MgC1; 40 mM CaCl2; 0.5 M sucrose; 2 mg/ml lysozyme; and 60 U/ml mutanolysin). All samples were incubated at 37 °C and the course of protoplast formation was followed by phase-contrast microscopy. After 2-h incubation, most cells had turned into protoplasts. The protoplasts of both strains were divided into three samples of 1 ml each. The first sample consisted of intact protoplasts. The protoplasts in the second sample were treated with trypsin (1 mg/ml, Sigma) and the protoplasts in the third sample were treated with a final concentration of 2% Triton X-100 before trypsin (1 mg/ml) treatment. All samples were incubated at 37 °C for 30 min. The intact and trypsin digested protoplasts were harvested by centrifugation (10,000×g, 5 min, 4 °C). The Triton X-100-treated samples were concentrated and an equal amount of each assay was boiled in SDS-PAGE sample buffer for 5 min before separation of proteins on an SDS polyacrylamide gel. The content of AmyQ in the four samples was analyzed by immunoblotting as described above.
Quantitation of cell lysis
Cell lysis was quantified by measuring the release of X-prolyl dipeptidyl aminopeptidase (PepX) from protoplasts. PepX activity in a standard volume of protoplast supernatant was determined by monitoring hydrolysis of the synthetic substrate Gly-Pro p-nitroanilide hydrochloride (Sigma) after 30 min incubation at 37 °C. The reaction was stopped by adding acetic acid to the samples (final concentration 24%) and the change in absorbance was monitored on a spectrophotometer (U-2000, Hitachi) at 410 nm.
Pulse-chase experiments
GM17 broth supplemented with 1% glucose, 5 μg/ml erythromycin, and chloramphenicol was inoculated with 3% aliquots of overnight cultures of L. lactis NZ9000(pKTH5041+pIL277) and NZ9000(pKTH5041+pKTH5066). The strains were propagated at 30 °C to an OD600=0.45 before 3 ng/ml nisin was added. After 1 h of induction the cells were harvested by centrifugation (2,000×g, 10 min at 4 °C), washed in CDM lacking methionine, resuspended in CDM without methionine, and incubated for 5 min at 30 °C. The methionine-deprived cultures were pulse-labeled for 2 min after addition of 20 μCi/ml of [35S]Met (ICN Biomedicals). A total of 1 ml 5% methionine was added (chase) and a 1.8-ml sample was taken immediately. Aliquots of 1.8 ml were withdrawn after 1, 2, 5, 10, and 15 min of chase and kept on ice water. All samples were centrifuged (18,000×g, 15 min) at 4 °C and the cells and the culture medium were precipitated by the addition of 10% tricarboxylic acid and washed with ice-cold acetone. The cell fractions were further resuspended in 400 μl of NET buffer (150 mM NaCI, 2 mM EDTA, and 20 mM Tris; pH 7.8), supplemented with 10 mg/ml lysozyme and 60 U/ml mutanolysin, and incubated at 37 °C for 1.5 h. For cell lysis, the samples were treated with SDS (final concentration 0.5%) and incubated at 95 °C for 5 min. Triton X-100 was added at a final concentration of 2%. The supernatant fractions were suspended in 120 μl NET with 0.5% SDS and diluted twofold by the addition of 120 μl NET with 4% Triton X-100. All samples were finally diluted with NET to a volume of 750 μl before immunoprecipitation.
Immunoprecipitation
Immunoprecipitation was carried out with a specific antibody against AmyQ bound to Protein A Sepharose beads (Amersham Pharmacia Biotech AB) as described by Bonifacino et al. (1999). The precipitated cell pellets were finally resuspended in 45 μl of NuPAGE® LDS Sample Buffer (Novex). Samples were centrifuged to remove the beads and boiled for 5 min before electrophoresis.
Results
Construction of strains
PrsA, AmyQ and PenP expressing L. lactis strains were constructed by transforming L. lactis NZ9000 (de Ruyter et al. 1996a) with plasmids pKTH5066, pKTH5041 and pKTH5054 respectively. To study the effect of PrsA on the secretion of AmyQ and PenP, pKTH5066 was introduced as a second plasmid into NZ9000(pKTH5041) and NZ9000(pKTH5054). Two control double-plasmid strains, lacking prsA, were also prepared by introducing the intact pIL277 instead of pKTH5066 into NZ9000(pKTH5041) and NZ9000(pKTH5054).
Production of PrsA in L. lactis
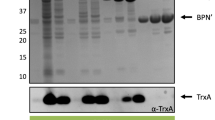
Production of PrsA in L. lactis was studied from the disrupted cells of nisin-induced L. lactis NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5054+pKTH5066) with Western blotting. With the three induction levels used, 0.1, 1, and 10 ng nisin/ml culture medium, no differences in the growth rate of the strains, determined by optical density measurement at 600 nm, could be observed (data not shown). Furthermore, the production level of PrsA was similar in both strains (data not shown). At the lowest induction level tested (0.1 ng nisin/ml culture medium) no PrsA could be detected in our experiments (data not shown). At the higher induction levels tested the PrsA protein appeared as a strong band on the Western blots (Fig. 1a), corresponding to the size of the mature PrsA produced in B. subtilis (Fig. 1b). The production of PrsA did not markedly differ at the induction levels of 1 and 10 ng nisin/ml culture medium but some differences could be found in the yield of PrsA as a function of growth (Fig. 1c). In the expression system used, the number of PrsA molecules per L. lactis cell (3×105) was 15-fold higher than that in the wild-type B. subtilis (2×104), and slightly higher than the number of PrsA molecules in a B. subtilis strain overexpressing PrsA (2×105, Fig. 1a; Vitikainen et al. 2001), suggesting that the amount of PrsA was most likely saturating for AmyQ production in L. lactis.

Production of PrsA in L. lactis NZ9000(pKTH5041+pKTH5066). a Western blot analysis and subsequent quantitation of PrsA protein bands of NZ9000(pKTH5041+pKTH5066) cell fractions (0.63–5 μl) after 5 h of induction with 10 ng nisin/ml culture medium (black bars) compared to a purified PrsA standard (5–50 ng of a nonlipomodified form of PrsA devoid of the diacylglycerol lipid anchor secreted from B. subtilis) (gray bars). b Comparison of PrsA produced in L. lactis NZ9000(pKTH5041+pKTH5066) and wild-type B. subtilis. Western analyses were performed with a rabbit antiserum against PrsA. c The amount of PrsA molecules on NZ9000(pKTH5041+pKTH5066) cells harvested at different time points after induction with 1 (open square) or 10 ng nisin/ml culture medium (filled square)
The effect of PrsA on the total yield of AmyQ
To investigate the effect of PrsA on AmyQ production in L. lactis the strains NZ9000(pKTH5041+pKTH5066), coexpressing amyQ and prsA, and NZ9000(pKTH5041+pIL277), expressing amyQ, were induced with different amounts of nisin (0.1, 1, and 10 ng/ml). During induction, aliquots of culture were withdrawn at the exponential and stationary phases of growth followed by Western blot analysis of the cell and supernatant fractions. With the lowest induction level (0.1 ng nisin/ml culture medium) no AmyQ could be detected with anti-AmyQ antibodies. However, an increase in the induction level resulted in distinct AmyQ bands on the blot (Fig. 2). Comparison of the two strains revealed that the presence of PrsA resulted in a substantial increase of AmyQ accumulation in both cell and supernatant fractions of NZ9000(pKTH5041+pKTH5066) (Fig. 2). The highest yield of AmyQ was obtained in early stationary phase (5 h after induction). At this time point the coproduction of PrsA resulted in a 25- to 30-fold increase of AmyQ in the cell fraction and a 7- to 9-fold increase of AmyQ in the supernatant fraction when 1 ng nisin/ml culture medium was used for induction (Fig. 2a,b). When the induction level was increased to 10 ng nisin/ml culture medium the amount of AmyQ increased two- to fourfold in the cell and supernatant fractions of both NZ9000(pKTH5041+pIL277) and NZ9000(pKTH5041+pKTH5066) (Fig. 2c,d). Thus, the difference in the amount of AmyQ remained essentially the same between the strains. The Western blots also revealed that essentially all cell-associated AmyQ was in the precursor form whereas the size of AmyQ in the culture medium corresponded to the size of processed mature AmyQ (Fig. 2). These results demonstrated that the presence of PrsA increased the yield of AmyQ in L. lactis, both as precursor in the cell fraction and as secreted AmyQ in the culture medium.

Production of AmyQ in induced L. lactis strains NZ9000(pKTH5041+pIL277) and NZ9000(pKTH5041+pKTH5066). Cell and supernatant fractions of NZ9000(pKTH5041+pKTH5066) (expressing amyQ and prsA) and NZ9000(pKTH5041+pIL277) (expressing amyQ) were induced with 1 ng nisin /ml culture medium (a, b) and 10 ng nisin /ml culture medium (c, d). Western blot analysis of samples withdrawn at different time points of induction was performed with rabbit antiserum against AmyQ. The bars represent the quantitation of the intensity of protein bands illustrated in Western blots
The effect of PrsA on AmyQ activity
The AmyQ activity was assayed from the same cell and supernatant fractions of induced NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5041+pIL277) strains that were used in the Western blot experiment. The activity assay showed that over 95% of AmyQ activity was located in the supernatant fractions both in L. lactis NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5041+pIL277) and that only a small amount of AmyQ activity was present in the cell fractions in both strains (data not shown). This result was in perfect agreement with the results from the Western experiment that showed that most cell-associated AmyQ was in its precursor form. With the induction level of 0.1 ng nisin/ml culture medium, no substantial difference in AmyQ activity in the supernatant fractions was obtained between NZ9000(pKTH5041+pKTH5066) and the control strain NZ9000(pKTH5041+pIL277) (Fig. 3a). However, when the induction level was increased, the difference in AmyQ activity in the supernatant fractions increased as a function of the induction time reaching its maximum after 5 h of induction (Fig. 3b). At this time point the prsA carrying strain NZ9000(pKTH5041+pKTH5066) secreted approximately sixfold more AmyQ activity to the culture medium than the control strain NZ9000(pKTH5041+pIL277). Induction levels over 1 ng nisin/ml culture medium resulted in only a small increase in AmyQ activity (Fig. 3c). After 7 h of induction the accumulation of AmyQ in the culture medium leveled off in both strains and the AmyQ activity in the supernatant remained stable even in O/N cultures (data not shown). These results indicated that the presence of PrsA resulted in a substantial increase in the yield of secreted, active AmyQ in the culture medium.

The amount of active AmyQ and PenP in the culture medium of induced L. lactis strains at different time points after addition of nisin. L. lactis NZ9000(pKTH5041+pKTH5066) (filled square) and NZ9000(pKTH5041+pIL277) (open square) were induced with a 0.1, b 1, and c 10 ng nisin/ml culture medium. The quantities of AmyQ were calculated from a standard curve drawn according to the activity of a purified AmyQ standard. d The activity of PenP in the supernatant fraction of L. lactis NZ9000(pKTH5054+pKTH5066) (filled square) and NZ9000(pKTH5054+pIL277) (open square) as measured after 1.5–7 h of induction with 10 ng nisin /ml culture medium
The effect of PrsA on production of PenP
When penP was expressed under the same induction conditions as described for amyQ, activity assays on the cell and supernatant fractions demonstrated that over 98% of PenP activity was found in the supernatant fraction and that the secreted PenP activity was on the same level in the presence and absence of PrsA (Fig. 3d). Western blot experiments demonstrated that approximately 50% of synthesized PenP remained cell-bound in both strains and there was no difference in the amount of PenP precursor between the two strains (data not shown). These results suggested that PenP was not a target protein for PrsA in L. lactis, a result that is in perfect agreement with PenP studies on B. subtilis (Kontinen and Sarvas 1988; Vitikainen et al. 2005).
Accumulation of pre-AmyQ on the outer surface of the cytoplasmic membrane
To investigate the location of pre-AmyQ in the cell fractions, protoplasts were prepared from the amyQ-expressing strain NZ9000(pKTH5041+pIL277) and amyQ and prsA-expressing strain NZ9000(pKTH5041+pKTH5066) induced either with 1 or 10 ng nisin/ml culture medium. PepX was used as an intracellular marker enzyme to quantify the proportion of disrupted protoplasts during their preparation. In our experiments, 5 to 10% of the total PepX activity could be detected in the protoplast supernatant fractions (results not shown), indicating that over 90% of the protoplasts remained intact. To define the location of AmyQ in the protoplasts, various treatments were carried out on the cells followed by trypsin digestion and immunoblotting analysis using antibodies against AmyQ. In the intact cells of NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5041+pIL277), disrupted with a cell mill, and in their protoplasts, which were not treated with trypsin, pre-AmyQ was found as a distinct band on Western blots at both induction levels (Fig. 4). From intact protoplasts, digested with trypsin, only trace amounts of AmyQ was detected at both induction levels (Fig. 4), suggesting that most of the cell-associated AmyQ was translocated through the cell membrane and was located on the trans side of the membrane. From protoplast samples, treated with trypsin after addition of Triton X-100, no detectable bands were found on the Western blots even after overexposure of the blots detected with a chemiluminiscence substrate (Fig. 4). In the control strain NZ9000(pKTH5041+pIL277), the amount of AmyQ was, however, substantially lower than in the PrsA-expressing strain NZ9000(pKTH5041+pKTH5066) (Fig. 4). These results were in agreement with the results of the Western blot experiment.

Western blot analysis of intact cells, protoplasts, and trypsin-digested protoplasts of L. lactis NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5041+pIL277) after 5 h of induction with 1 or 10 ng nisin/ml culture medium. A specific rabbit antiserum against AmyQ was used for detection. c Cells and p protoplasts
Kinetics of pre-AmyQ processing in L. lactis NZ9000(pKTH5041+pIL277) and NZ9000(pKTH5041+pKTH5066)
Processing of the precursor amylase in prsA [NZ9000(pKTH5041+pKTH5066)] and non-prsA [NZ9000(pKTH5041+pIL277)] expressing L. lactis strains was analyzed by pulse-chase labeling experiments using [35S]Met and immunoprecipitation. During the 15-min chase, the processing of preamylase to mature enzyme was relatively slow in both strains (T/2 approximately 3 min). In the amyQ- and prsA-expressing strain NZ9000(pKTH5041+pKTH5066), the proportion of labeled mature AmyQ increased from 30 to 75% during 5 min and remained at the same level after 15 min of chase (Fig. 5). In the control strain NZ9000(pKTH5041+pIL277), the intensity of labeled AmyQ was 4–5 times lower than that of NZ9000(pKTH5041+pKTH5066) during the entire chase (Fig. 5), but no substantial differences in the processing kinetics could be found. These results strongly suggested that PrsA increased the yield of AmyQ in the L. lactis strain coexpressing amyQ and prsA and did not affect the rate of translocation or processing of pre-AmyQ.

Maturation and secretion kinetics of AmyQ in induced L. lactis NZ9000(pKTH5041+pKTH5066) and NZ9000(pKTH5041+pIL277) studied by pulse-labeling. Radiolabeled samples were immunoprecipitated using rabbit antiserum against AmyQ and AmyQ bands were illustrated on LDS-PAGE gels. p Precursor AmyQ and m mature AmyQ
Discussion
Efforts have previously been made to enhance the secretion efficiency of heterologous proteins in L. lactis, e.g., by exploration of different signal peptides and modifications of regions downstream of the signal peptide cleavage site (for references, see Le Loir et al. 2005). The fusion of labile proteins to stable carriers and inactivation of protease genes in the production host are other methods that have been used to increase the yield of exported proteins in L. lactis (for references, see Le Loir et al. 2005). Still, the components of the lactococcal secretion apparatus and their role in the secretion process are characterized only to a very limited extent. In this work, we have studied the effect of the extracellular chaperone-like protein PrsA of B. subtilis on the secretion of heterologous proteins in L. lactis. The B. amyloliquefaciens AmyQ and the B.licheniformis PenP were chosen as model proteins due to their different dependency on PrsA in B. subtilis.
When expressed from an inducible expression system we found that the PrsA production level (3×105 molecules/cell) in L. lactis is comparable to, or even exceeds, that maximally obtained in B. subtilis with a multicopy expression system (2×105 molecules/cell) (Vitikainen et al. 2001). The molecular size of PrsA indicated that PrsA was processed and folded into its functional form in L. lactis.
The lack of choice of available inducible promoter units in L. lactis resulted in the use of the PnisA promoter in all our expression constructs, thus, hampering an independent expression control of the test components. Even though PrsA was partly degraded during growth, the amount of PrsA molecules would most likely be enough for AmyQ secretion in our study. On the other hand, the high amount of PrsA did not hamper the growth of cells nor did it affect the secretion process and kinetics of the nontarget protein PenP, the secretion of which was indistinguishable in the presence and absence of PrsA. Furthermore, there was no difference in the amount of PrsA produced by the AmyQ- and PenP-producing strains, indicating that the nature of the reporter gene did not affect the expression of prsA.
Compared to the control strain lacking PrsA, the coproduction of AmyQ and PrsA increased the amount of AmyQ up to 15- to 20-fold at the maximum induction level used. At the highest induction level hardly any AmyQ, detected on Western blots, was located in the cytoplasm, indicating an efficient translocation of the protein through the cytoplasmic membrane. The small amount of AmyQ precursor observed in the protoplast fraction, even after trypsin treatment, could either represent precursor molecules located in the cytoplasm, within the cytoplasmic membrane or on the protoplast surface where they could be protected from trypsin degradation by the remnants of the cell wall material.
Although the presence of PrsA resulted in considerable accumulation of translocated cell-associated pre-AmyQ, only part of the precursor molecules were correctly processed and folded into mature AmyQ molecules that were secreted to the culture medium. This may suggest that the cleavage of the signal peptide of pre-AmyQ could be one bottleneck for more efficient secretion of AmyQ. Still, the presence of PrsA resulted in a significant increase (sixfold) in AmyQ activity in the culture medium of L. lactis.
A surprising finding regarding AmyQ activity in the supernatant fraction was the apparent discrepancy between AmyQ activity and AmyQ cross-reacting material when the induction level was raised from 1 to 10 ng nisin/ml culture medium. It is clear that Western blot assays are notoriously inaccurate when small differences are compared but the observed two- to threefold difference seemed to be too consistent to be regarded just as an assay error. Thus, the lack of other obvious alternatives makes us consider that at a high expression level, part of the processed and secreted AmyQ molecules are not fully active and thus, probably not correctly folded despite the presence of PrsA.
In all our experiments we could see an increased amount of cell-associated and secreted AmyQ in the prsA strain when compared to the control strain lacking prsA. It is not clear how PrsA performs its role in increasing the amount of AmyQ in L. lactis. In B. subtilis PrsA was suggested to have a chaperone-like function in protecting newly translocated proteins from degradation by extracellular proteases by preventing interactions of the translocated nascent protein with components on the cytoplasmic membrane and in the cell wall (Colomer-Pallas et al. 2004; Wahlstrom et al. 2003). We could not observe any AmyQ degradation products in our Western blot experiments. Similar results have, however, been observed earlier in Western blot experiments with AmyQ produced in B. subtilis (Kontinen and Sarvas 1988). It is feasible that when the correct folding of AmyQ is compromised, the unfolded or partially folded proteins are exposed to proteolysis and become rapidly degraded into small fragments, which are unrecognizable to AmyQ antiserum. Another explanation to the positive effect of PrsA on AmyQ production could be that PrsA plays a role in the transcription of AmyQ (but not PenP). However, no such function has ever been observed for PrsA. Furthermore, overproduction of L. lactis PmpA, a PrsA-like protein identified in L. lactis, was found to prevent degradation of proteins on the cell surface of L. lactis (Drouault et al. 2002). Considering these results we find it most likely that the function of PrsA on AmyQ in L. lactis is very similar to its function in B. subtilis in protecting its target proteins from degradation by cell wall-associated proteases.
Studies in B. subtilis, the model organism of protein secretion in gram-positive bacteria, suggest that the problems in secretion of heterologous proteins are often associated with the events after translocation, like signal processing, refolding, cell wall interactions, and degradation of the protein product (Bolhuis et al. 1999). In this work we demonstrated that B. subtilis PrsA executed a chaperone-like function on exported proteins also in a heterologous environment. It is clear that the use of PrsA is limited to its target proteins but the characterization of PmpA in L. lactis indicates that extracellular chaperones are also present in other gram-positive bacteria. One might also speculate that the substrate profiles of different extracellular chaperones are probably different. It would therefore be of great value to characterize and further develop different extracellular chaperones that could be used in the bacterial production of industrially and pharmaceutically important proteins.
References
Bermudez-Humaran LG, Langella P, Commissaire J, Gilbert S, Le Loir Y, L’Haridon R, Corthier G (2003) Controlled intra- or extracellular production of staphylococcal nuclease and ovine omega interferon in Lactococcus lactis. FEMS Microbiol Lett 224:307–313
Bolhuis A, Tjalsma H, Smith HE, de Jong A, Meima R, Venema G, Bron S, van Dijl JM (1999) Evaluation of bottlenecks in the late stages of protein secretion in Bacillus subtilis. Appl Environ Microbiol 65:2934–2941
Bonifacino JS, Dell’Angelica EC, Springer TA (eds) (1999) Analysis of proteins. Immunoprecipitation. Green Publishing Associates, New York, pp 10.16.1–10.16.29
Colomer-Pallas A, Petit-Glatron MF, Chambert R (2004) Bacillus subtilis alpha-amylase: interactions of a partially folded conformer with small unilamellar vesicles. Biochim Biophys Acta 1660:16–23
de Ruyter PG, Kuipers OP, Beerthuyzen MM, van Alen-Boerrigter I, de Vos WM (1996a) Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J Bacteriol 178:3434–3439
de Ruyter PG, Kuipers OP, de Vos WM (1996b) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62:3662–3667
Drouault S, Anba J, Bonneau S, Bolotin A, Ehrlich SD, Renault P (2002) The peptidyl-prolyl isomerase motif is lacking in PmpA, the PrsA-like protein involved in the secretion machinery of Lactococcus lactis. Appl Environ Microbiol 68:3932–3942
Holo H, Nes IF (1995) Transformation of Lactococcus by electroporation. Methods Mol Biol 47:195–199
Kontinen VP, Sarvas M (1988) Mutants of Bacillus subtilis defective in protein export. J Gen Microbiol 134:2333–2344
Kontinen VP, Sarvas M (1993) The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol Microbiol 8:727–737
Kontinen VP, Saris P, Sarvas M (1991) A gene (prsA) of Bacillus subtilis involved in a novel, late stage of protein export. Mol Microbiol 5:1273–1283
Le Loir Y, Azevedo V, Oliveira SC, Freitas DA, Miyoshi A, Bermudez-Humaran LG, Nouaille S, Ribeiro LA, Leclercq S, Gabriel JE, Guimaraes VD, Oliveira MN, Charlier C, Gautier M, Langella P (2005) Protein secretion in Lactococcus lactis: an efficient way to increase the overall heterologous protein production. Microb Cell Fact 4:2
Leskela S, Wahlstrom E, Kontinen VP, Sarvas M (1999) Lipid modification of prelipoproteins is dispensable for growth but essential for efficient protein secretion in Bacillus subtilis: characterization of the lgt gene. Mol Microbiol 31:1075–1085
Lindholm A, Smeds A, Palva A (2004) Receptor binding domain of Escherichia coli F18 fimbrial adhesin FedF can be both efficiently secreted and surface displayed in a functional form in Lactococcus lactis. Appl Environ Microbiol 70:2061–2071
Neugebauer K, Sprengel R, Schaller H (1981) Penicillinase from Bacillus licheniformis: nucleotide sequence of the gene and implications for the biosynthesis of a secretory protein in a gram-positive bacterium. Nucleic Acids Res 9:2577–2588
Novotny R, Scheberl A, Giry-Laterriere M, Messner P, Schaffer C (2005) Gene cloning, functional expression and secretion of the S-layer protein SgsE from Geobacillus stearothermophilus NRS 2004/3a in Lactococcus lactis. FEMS Microbiol Lett 242:27–35
Otto R, Brink Bt, Veldkamp H, Konings WN (1983) The relation between growth rate and electrochemical proton gradient of Streptococcus cremoris. FEMS Microbiol Lett 16:69–74
Palva I (1982) Molecular cloning of alpha-amylase gene from Bacillus amyloliquefaciens and its expression in B. subtilis. Gene 19:81–87
Poolman B, Konings WN (1988) Relation of growth of Streptococcus lactis and Streptococcus cremoris to amino acid transport. J Bacteriol 170:700–707
Sarvas M, Harwood CR, Bron S, van Dijl JM (2004) Post-translocational folding of secretory proteins in gram-positive bacteria. Biochim Biophys Acta 1694:311–327
Savijoki K, Kahala M, Palva A (1997) High level heterologous protein production in Lactococcus and Lactobacillus using a new secretion system based on the Lactobacillus brevis S-layer signals. Gene 186:255–262
Simon D, Chopin A (1988) Construction of a vector plasmid family and its use for molecular cloning in Streptococcus lactis. Biochimie 70:559–566
Simons K, Sarvas M, Garoff H, Helenius A (1978) Membrane-bound and secreted forms of penicillinase from Bacillus licheniformis. J Mol Biol 126:673–690
Takkinen K, Pettersson RF, Kalkkinen N, Palva I, Soderlund H, Kaariainen L (1983) Amino acid sequence of alpha-amylase from Bacillus amyloliquefaciens deduced from the nucleotide sequence of the cloned gene. J Biol Chem 258:1007–1013
Vitikainen M, Pummi T, Airaksinen U, Wahlstrom E, Wu H, Sarvas M, Kontinen VP (2001) Quantitation of the capacity of the secretion apparatus and requirement for PrsA in growth and secretion of alpha-amylase in Bacillus subtilis. J Bacteriol 183:1881–1890
Vitikainen M, Hyyrylainen HL, Kivimaki A, Kontinen VP, Sarvas M (2005) Secretion of heterologous proteins in Bacillus subtilis can be improved by engineering cell components affecting post-translocational protein folding and degradation. J Appl Microbiol 99:363-375
Wahlstrom E, Vitikainen M, Kontinen VP, Sarvas M (2003) The extracytoplasmic folding factor PrsA is required for protein secretion only in the presence of the cell wall in Bacillus subtilis. Microbiology 149:569–577
Acknowledgements
This work was supported by the Academy of Finland (57909). We thank Dr. Ilkka Palva for valuable discussions and critical reading of the manuscript and for providing the specific antibody against AmyQ. We also thank Dr. Matti Sarvas for valuable discussions and Dr. Vesa Kontinen for providing the purified PrsA protein and the anti-PrsA and anti-PenP serums. Finally, we thank Anja Osola for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lindholm, A., Ellmén, U., Tolonen-Martikainen, M. et al. Heterologous protein secretion in Lactococcus lactis is enhanced by the Bacillus subtilis chaperone-like protein PrsA. Appl Microbiol Biotechnol 73, 904–914 (2006). https://doi.org/10.1007/s00253-006-0551-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0551-y