
Abstract
Pollutants are frequently found as mixtures yet it is difficult to engineer enzymes with broad substrate ranges on aromatics. Inspired by the archetypal nitroarene dioxygenase, which shares its electron transport with a salicylate monooxygenase, we have created an innovative and general approach to expand the substrate range of dioxygenase enzymes in a single cell. We have developed here a series of novel, hybrid dioxygenase enzymes that function with a single ferredoxin reductase and ferredoxin that are used to transport two electrons from nicotinamide adenine dinucleotide to the two independent terminal oxygenases. Each independent alpha-oxygenase may then be used simultaneously to create orthric enzymes that degrade mixtures of environmental pollutants. Specifically, we created a hybrid dioxygenase system consisting of naphthalene dioxygenase/dinitrotoluene dioxygenase to simultaneously degrade 2,4-dinitrotoluene and naphthalene (neither enzyme alone had significant activity on both compounds) and dinitrotoluene dioxygenase/nitrobenzene dioxygenase to simultaneously degrade the frequently encountered 2,4,6-trinitrotoluene reduction products 2-amino-4,6-dinitrotoluene and 4-amino-2,6-dinitrotoluene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
2,4-Dinitrotoluene (24DNT) is a priority pollutant (Keith and Telliard 1979) used during the synthesis of the explosive 2,4,6-trinitrotoluene (TNT) (Spanggord et al. 1991), polyurethane foams, toluene diisocyanate, and dyestuffs (Howe-Grant 1991). Naphthalene is also a priority pollutant (Keith and Telliard 1979) that is generated during the refining of crude oil and used in the synthesis of phthalic anhydride (Howe-Grant 1991). Both pollutants pose significant health threats when ingested by mammals (Mori et al. 2000; Stohs et al. 2002). Under environmental conditions, TNT is easily reduced to the aminodinitrotoluene isomers 2-amino-4,6-dinitrotoluene (2A46DNT) and 4-amino-2,6-dinitrotoluene (4A26DNT) (Fig. 1) (Yin et al. 2005); these reduction products were reported to be more toxic than the parent compound TNT (Johnson et al. 1994). According to the US Environmental Protection Agency, a majority of superfund sites contain a mixture of pollutants, and various sites contain mixtures of 24DNT and naphthalene (Green 1998) and mixtures of 24DNT, 2A46DNT, 4A26DNT, 4-nitrotoluene (4NT), and naphthalene (ATSDR 2004). There has been little work focused on degrading simultaneous mixtures of these structurally diverse compounds.

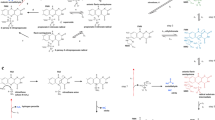
Wild-type dioxygenase reactions with 2-amino-4,6-dinitrotoluene, 4-amino-2,6-dinitrotoluene, naphthalene, 2,4-dinitrotoluene, and 4-nitrotoluene and the monooxygenase-like reactions toward 2-amino-4,6-dinitrotoluene and 4-nitrotoluene. The wild-type dioxygenase system with the highest rate of product formation is denoted above each reaction arrow
Recombinant Escherichia coli strains expressing 24DNT dioxygenase (R34DDO) from Burkholderia cepacia R34 and Burkholderia sp. strain DNT (DNTDDO) are capable of oxidizing 2A46DNT at the 3,4 position and releasing nitrite to form 3-amino-4-methyl-5-nitrocatechol (3A4M5NC) and hydroxylating the methyl group to form 2-amino-4,6-dinitrobenzyl alcohol (2A46DNBA) (Fig. 1) (Johnson et al. 2001). The recombinant nitrobenzene dioxygenase (NBDO) from Comamonas sp. strain JS765 was shown to oxidize 4A26DNT at the 2,3 position releasing nitrite and to generate 3A6M5NC (Fig. 1) (Johnson et al. 2001). The naphthalene dioxygenase (NDO) from Ralstonia sp. strain U2 generates (1R, 2S)-cis-1,2-dihydro-1,2-dihydroxynaphthalene (naphthalenediol) from naphthalene (Fuenmayor et al. 1998) (Fig. 1) and was reported to have no activity toward 24DNT, 2A46DNT, or 4A26DNT (Keenan et al. 2005). We sought to take advantage of these various activities by creating efficient hybrid aromatic dioxygenases (Table 1 and Fig. 2).

Orthric NDO-R34DDO (two terminal oxygenases sharing the same electron transfer subunits) in which NDO NagAa (ferredoxin reductase) and NagAb (ferredoxin) transfer electrons to both the alpha oxygenase subunit NDO NagAc and the alpha oxygenase subunit R34DDO DntAc for the oxidation of both naphthalene to (1R, 2S)-cis-1,2-dihydro-1,2-dihydroxynaphthalene (naphthalenediol) and 2,4-dyinitrotoluene to 4-methyl-5-nitrocatechol, respectively
To generate variant enzyme systems that are equally active toward many structurally diverse substrates, our goal was to engineer novel dioxygenase constructs containing two terminal oxygenase components with different catalytic capabilities. Replacing just the alpha-subunit from the Rieske biphenyl dioxygenase (BPDO) of B. cepacia LB400 with the BPDO alpha-subunit from Pseudomonas pseudoalcaligenes KF707 led to novel activity of KF707 BPDO toward 2,2′chlorobiphenyl; however, this hybrid BPDO system lost wild-type KF707 BPDO activity toward 4,4′-chlorobiphenyl and 2,5,4′-chlorobiphenyl (Kumamaru et al. 1998). The terminal oxygenase alpha-subunit controls substrate specificity for the biphenyl (Erickson and Mondello 1993) and NDO (Parales et al. 2000a), and exchanges between the alpha-subunits and beta-subunit from closely related naphthalene and nitroarene dioxygenases systems can inhibit dioxygenase activity (Parales et al. 1998a,b). When the alpha-subunit of the 2-nitrotoluene dioxygenase (2NTDO) from Acidovorax sp. strain JS42 was combined with the beta-subunit of the DNTDDO, the hybrid enzyme had threefold lower activity toward 2-nitrotoluene, relative to 2NTDO, and no activity toward 24DNT, relative to DNTDDO (Parales et al. 1998a). Similarly, when the alpha-subunit of the NDO (NCIB NDO) from Pseudomonas sp. strain NCIB 9816-4 was combined with the beta-subunit of either 2NTDO, DNTDDO, or the toluene dioxygenase from Pseudomonas putida F1, the hybrid NCIB NDO enzymes had fourfold lower, eightfold lower, and no activity toward naphthalene relative to the wild-type NCIB NDO, respectively (Parales et al. 1998b). Therefore, it is our contention that hybrid dioxygenase systems should include both the alpha-subunit and beta-subunit from the same dioxygenase system to maintain a broad range of catalytic activity, allowing functional associations between terminal dioxygenase components, and the rates of substrate oxidation will not be decreased relative to the individual wild-type dioxygenase systems. However, the electron transfer subunits may be shared.
Our constructs here are inspired by the Ralstonia sp. strain U2 NDO (NagAaAbAcAd) where the NagAa (ferredoxin reductase) and NagAb (ferredoxin) components are shared with the salicylate 5-hydroxylase (NagAaGHAb) (Fuenmayor et al. 1998) to allow the catabolism of naphthalene through gentisate rather than through catechol (Fuenmayor et al. 1998). The compatibility of NagAb with both oxygenase alpha-subunits, NagG, and NagAc, is astounding considering that these two proteins share only 20% amino acid similarity. Note the ferredoxin component is the bridge between nicotinamide adenine dinucleotide (NADH) and the terminal oxygenase (Fig. 2) (Lee 1998). Remnants of nagG-like genes were identified in the R34DDO, DNTDDO, and NBDO dioxygenase loci, which provides strong evidence for the evolutionary relationship between these nitroarene dioxygenases and NDO (Fuenmayor et al. 1998; Johnson et al. 2002; Lessner et al. 2002; Suen et al. 1996), and which suggests these dioxygenase subunits may also be made to work with a single ferredoxin reductase and ferredoxin while each alpha-subunit may be used for oxidizing different substrates (Fig. 2). Similar to Orthrus, the two-headed dog from Greek mythology (Gantz 1993), we have devised novel hybrid dioxygenase systems where two heads (two independent alpha/beta terminal dioxygenases) are better than one (Table 1 and Fig. 2) (note that chimera implies three different heads, but a single animal; Gantz 1993). Using the NagAaAbAcAd NDO system from strain U2, it was determined that the native E. coli host ferredoxin could not supply the NDO terminal oxygenase with the electrons needed for the oxidation of salicylate (Fuenmayor et al. 1998). The deletion of the nagAb gene from the nagA loci resulted in loss of salicylate oxidation by the E. coli strain carrying the wild-type U2 nagAa, nagAc, and nagAd genes; therefore, it is necessary to include the ferredoxin reductase and ferredoxin genes from a Rieske dioxygenase system (Fuenmayor et al. 1998). Through dioxygenase loci engineering, we generated the orthric dioxygenase systems (1) NDO-R34DDO (Fig. 2), which simultaneously oxidized 24DNT and naphthalene as well as 2A46DNT or 4-nitrotoluene (4NT); (2) R34DDO-NBDO and NDO-NBDO-DNTDDO, which simultaneously oxidized 2A46DNT and 4A26DNT; and (3) NDO-NBDO, which simultaneously oxidized naphthalene and 4A26DNT as well as 4NT. This is the first report of using two separate terminal oxygenases with shared electron transport to expand enzyme activity, to describe a dioxygenase system capable of simultaneously degrading mixtures of 24DNT and naphthalene and mixtures of 2A46DNT and 4A26DNT, and to investigate the compatibility of the ferredoxin subunit from a Rieske nonheme iron dioxygenase with a dual terminal dioxygenase system.
Materials and methods
Bacterial strains, growth conditions, and protein gel electrophoresis
E. coli strain TG1 (Sambrook et al. 1989) was used as the host for the recombinant dioxygenase constructs during the whole-cell transformations of 24DNT and naphthalene. E. coli JVQ2 with two nitroreductase mutations (nfsAB) (Whiteway et al. 1998) was used during whole-cell transformations of 2A46DNT, 4A26DNT, and 4NT to minimize the undesired reduction of the nitro groups.
For whole-cell dioxygenase transformations, single colonies were initially inoculated in 25 ml Luria–Bertani medium (LB) (Sambrook et al. 1989) containing 1% glucose and kanamycin (100 μg/ml) and grown overnight at 37 °C at 250 rpm. Glucose maintained plasmid segregational stability by suppressing dioxygenase expression until enzyme activity was desired. Ten milliliters of the overnight culture was inoculated in 250 ml LB media containing 1 mM isopropyl-β-d-thiogalactopyranoside and kanamycin (100 μg/ml) and grown at 37 °C at 250 rpm from an initial optical density (OD) at 600 nm of 0.3 to 2.0 (∼3 h incubation). R34DDO and DNTDDO were expressed in E. coli using plasmids pBS(Kan)R34 (Keenan et al. 2004) and pBS(Kan)DNT (Leungsakul et al. 2005); NDO was expressed using pBS(Kan)NDO− (Keenan et al. 2005); and NBDO was expressed using pBS(Kan)NBDO (Table 1). Due to the potential for an active salicylate 5-hydroxylase to interfere with the activity of the NDO, the nagGH genes were removed from the plasmid pBS(Kan)NDO using naturally occurring BsrGI restriction sites to generate pBS(Kan)NDO− as described previously (Keenan et al. 2005). The relative expression of R34DDO, NDO, NBDO, DNTDDO, NDO-R34DDO, R34DDO-NBDO, NBDO-DNTDDO, and NDO-NBDO-DNTDDO from E. coli was evaluated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Sambrook et al. 1989) with a 12% acrylamide gel.
Chemicals
Dimethyl formamide (DMF), naphthalene, 4-nitrobenzyl alcohol (4NBA), and 4-methylcatechol (4MC) were purchased from Fisher Scientific (Fairlawn, NJ, USA). 24DNT, 4A26DNT, 4NT, naphthalenediol, sulfanilamide (SUL), and N-(1-naphthyl)ethylenediamine dihydrochloride were purchased from Sigma Chemical (St. Louis, MO, USA). 2A46DNT was purchased from AccuStandard (New Haven, CT, USA). Product standards 4-methyl-5-nitrocatechol (4M5NC) and 3A4M5NC were supplied by Dr. Jim C. Spain of the US Air Force.
Construction of pBS(Kan)NBDO
As a positive control for the oxidation of 4A26DNT, pBS(Kan)NBDO (Fig. 3a) was constructed by polymerase chain reaction (PCR) amplification of the Comamonas sp. strain JS765 nbzAaAbAcAd loci from plasmid pDTG927 (Lessner et al. 2002), using standard PCR reaction conditions as described previously (Keenan et al. 2004) and the high-fidelity Pfu DNA polymerase. The front primer BSIINbzApaI forward (fwd) (Table 2) contains the restriction site ApaI and the rear primer BSIINbzSacI reverse (rvs) (Table 2) contains the restriction site SacI; these primers allowed the directional cloning of the 4-kb nbzAaAbAcAd loci downstream of the lac promoter of the expression vector pBS(Kan) [high copy number plasmid derived from pBluescript II KS(−)] (Canada et al. 2002) and generated the 8,068-bp plasmid pBS(Kan)NBDO. E. coli TG1 cells were transformed with the plasmid construct using BioRad Gene Pulser™ (Hercules, CA, USA) at 15 kV/cm, 25 μF, and 200 Ω.

Vectors a pBS(Kan)NBDO, b pBS(Kan)NDO-R34DDO, c pBS(Kan)R34DDO-NBDO, and d pBS(Kan)NDO-NBDO-DNTDDO for expression of wild-type and orthric dioxygenase systems. KanR is the kanamycin resistance gene. The Ralstonia sp. strain U2 naphthalene dioxygenase nagAc (alpha-subunit) and nagAd (beta-subunit) genes are shown in pBS(Kan)NDO− and pBS(Kan)NDO-R34DDO, whereas the nagAa (ferredoxin reductase) and nagAb (ferredoxin) genes are shown in pBS(Kan)NDO−, pBS(Kan)NDO-R34DDO, and pBS(Kan)NDO-NBDO-DNTDDO. The B. cepacia R34 2,4-dinitrotoluene dioxygenase dntAc (alpha-subunit) and dntAd (beta-subunit) genes are shown in pBS(Kan)NDO-R34DDO and pBS(Kan)R34DDO-NBDO, and the dntAa (ferredoxin reductase) and dntAb (ferredoxin) genes are shown in pBS(Kan)R34DDO-NBDO. The Comamonas sp. strain JS765 nitrobenzene dioxygenase nbzAc (alpha-subunit) and nbzAd (beta-subunit) genes are shown in pBS(Kan)R34DDO-NBDO and pBS(Kan)NDO-NBDO-DNTDDO. The Burkholderia sp. strain DNT 2,4-dinitrotoluene dioxygenase dntAc (alpha-subunit) and dntAd (beta-subunit) genes are shown in pBS(Kan)NDO-NBDO-DNTDDO
Construction of the orthric dioxygenase systems
To construct an orthric dioxygenase system capable of degrading both 24DNT and naphthalene, NDO-R34DDO, the 2-kb R34DDO dntAcAd terminal oxygenase genes, were amplified from pBS(Kan)R34 (Keenan et al. 2004) using Deep Vent high-fidelity DNA Polymerase (New England Biolabs, Beverly, MA, USA), the front primer R34 BsrGI fwd (Table 2), and the rear primer R34 BsrGI rvs (Table 2). Both primers contained the restriction site BsrGI for ligation into the naturally occurring BsrGI restriction site in pBS(Kan)NDO− downstream of the stop codon of nagAa and upstream of the start codon for nagAb (Fig. 3b). Because the BsrGI restriction site is located after the start codon of the disrupted nagG salicylate 5-hydroxylase alpha-subunit gene, the front primer R34 BsrGI fwd contained the in-frame stop codon TGA (Table 2) to prevent translation of the truncated NagG. After transformation into E. coli TG1, the correctly constructed plasmid was selected based on the production of indigoid compounds that are generated in E. coli from indole derived from tryptophan (Rui et al. 2005), and through the use of the 96-well nitrite assay after incubation with 24DNT. The constructed plasmid pBS(Kan)NDO-R34DDO is 10,076 bp.
To generate an orthric dioxygenase capable of 2A46DNT and 4A26DNT degradation, R34DDO-NBDO, the 2-kb NBDO nbzAcAd PCR product, was amplified using Nbz BsrGI fwd and Nbz BsrGI rvs (Table 2) and was ligated into pBS(Kan)R34 using the naturally occurring restriction site BsrGI downstream of the dntAa stop codon and upstream of the dntAb start codon generating the 10,095-bp plasmid pBS(Kan)R34DDO-NBDO (Fig. 3c). The ligation of the nbzAcAd fragment into the BsrGI restriction site of pBS(Kan)R34 removed the 1.2-kb open reading frame (ORF)1a and ORF1b truncated gene sequences. After transformation into E. coli TG1, the correctly constructed plasmid was selected based on the production of indigoid compounds from recombinant TG1 colonies and through the use of the 96-well nitrite assay after incubation with 4A26DNT.
The orthric dioxygenase plasmid pBS(Kan)NDO-NBDO-DNTDDO (Fig. 3d) was constructed in two steps to increase the oxidation of both 2A46DNT and 4A26DNT and to create an enzyme that simultaneously degrades 4A26DNT and naphthalene. First, the 2-kb NBDO nbzAcAd terminal oxygenase genes were amplified from pBS(Kan)NBDO using Deep Vent high-fidelity DNA Polymerase, the front primer Nbz BsrGI fwd (Table 2), and the rear primer Nbz BsrGI rvs (Table 2). Both primers contained the restriction site BsrGI, and the front primer contained an in-frame stop codon TGA (Table 2). The NBDO nbzAcAd PCR product was ligated into pBS(Kan)NDO− similar to the construction of pBS(Kan)NDO-R34DDO, generating the 10,072-bp plasmid pBS(Kan)NDO-NBDO. The plasmid pBS(Kan)NDO-NBDO was selected based on the production of indigoid compounds from recombinant TG1 colonies and through the use of the 96-well nitrite assay after incubation with 4A26DNT. Then the 2-kb DNTDDO dntAcAd terminal oxygenase genes were amplified using high-fidelity Pfu DNA Polymerase, the front primer DNT chimera Eco fwd (Table 2) (which contained the restriction site EcoRI), and the rear primer pBSii Back PCR rvs (Table 2) (which was downstream of the restriction site XbaI); these primers allowed the directional cloning of the 2-kb dntAcAd terminal oxygenase genes replacing the U2 nagAcAd genes (2 kb) and generated the 10,107-bp plasmid pBS(Kan)NDO-NBDO-DNTDDO. After transformation into E. coli TG1, the correctly constructed plasmid was selected based on the production of indigoid compounds from recombinant TG1 colonies.
The orthric dioxygenase plasmid pBS(Kan)NBDO-DNTDDO was constructed with the goal of increasing the activity of a NBDO toward 2A46DNT by amplifying the 2-kb DNTDDO dntAcAd terminal oxygenase gene product from pBS(Kan)DNT using high-fidelity Pfu DNA Polymerase, the front primer R34 BsrGI fwd, and the rear primer DNT BsrGI rvs (Table 2). Both primers contain the restriction site BsrGI, and the front primer contained the engineered in-frame stop codon. The DNT dntAcAd PCR product was ligated into the naturally occurring BsrGI site in pBS(Kan)NBDO downstream of the nbzAa stop codon and upstream of the nbzAb start codon, disrupted ORF2, and generated the 10,066-bp plasmid pBS(Kan)NBDO-DNTDDO. After transformation into E. coli TG1, the correctly constructed plasmid produced a very low amount of indigoid compounds from recombinant TG1 colonies, and the use of the 96-well nitrite assay after contact with 2A46DNT was critical.
To confirm the correct construction of the plasmids pBS(Kan)NBDO, pBS(Kan)NDO-R34DDO, pBS(Kan)R34DDO-NBDO, pBS(Kan)NDO-NBDO, pBS(Kan)NBDO-DNTDDO, and pBS(Kan)NDO-NBDO-DNTDDO, plasmid DNA was isolated using QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA, USA) and checked with the restriction enzymes AccI, ApaI, BamHI, BsrGI, Bsu36I, EcoRI, HindIII, KpnI, SacI, and XbaI (New England Biolabs). Once the correct plasmid construction was identified, the plasmid DNA was transformed into E. coli JVQ2.
To further confirm the construction of the correct plasmids, the 5′ and 3′ junctions between the NBDO nbzA loci and the pBS(Kan) cloning vector were sequenced as described previously (Keenan et al. 2004), along with the orthric dioxygenase insert junctions at each BsrGI cloning site of pBS(Kan)NDO-R34DDO, pBS(Kan)R34DDO-NBDO, pBS(Kan)NDO-NBDO, and pBS(Kan)NBDO-DNTDDO, and the EcoRI and XbaI cloning sites of pBS(Kan)NDO-NBDO-DNTDDO. The pBS(Kan)NBDO junctions were sequenced using the primers pBSKan junction fwd and pBSKan junction rvs (Table 2). The pBS(Kan)NDO-R34DDO and pBS(Kan)NDO-NBDO BsrGI junctions and the 5′ in-frame stop codon were sequenced using U2 Bsr junction fwd and U2 Bsr junction rvs (Table 2). The pBS(Kan)R34DDO-NBDO BsrGI junctions and the 5′ in-frame stop codon were sequenced using U2 Bsr junction fwd and R34 Bsr junction rvs (Table 2). The pBS(Kan)NBDO-DNTDDO BsrGI junctions and the 5′ in-frame stop codon were sequenced using U2 Bsr junction fwd and Nbz Bsr junction rvs (Table 2). The pBS(Kan)NDO-NBDO-DNTDDO 5′ EcoRI and 3′ XbaI junctions were sequenced using DNT Eco junction fwd and pBSKan junction rvs, respectively (Table 2). This showed the in-frame stop codons and oxygenase gene sequences were as expected, therefore no errors were introduced during PCR amplification of the terminal oxygenase genes. Through sequencing it was also verified that the ORF present in the R34DDO dntA loci (ORF1a and ORF2, Table 1) and in the NBDO nbzA loci (ORF2, Table 1) were truncated as desired in the final plasmid constructs pBS(Kan)R34DDO-NBDO and pBS(Kan)NBDO-DNTDDO, respectively.
Product identification and rates of formation through liquid chromatography
After constructing the wild-type and orthric dioxygenases, the activities of these systems were analyzed during whole-cell transformations using reverse-phase high-pressure liquid chromatography (HPLC) with a Chromolith Performance RP-18e column (Merck KGaA, Darmstadt, Germany) as described previously (Keenan et al. 2005). For all the substrates, 25 ml of exponentially grown cells were resuspended in 100 mM sodium phosphate buffer, pH 6.5, and were incubated in 250-ml glass shake flasks with 0.1 mM of 24DNT, naphthalene, or 4NT or 0.25 mM of 2A46DNT or 4A26DNT (DMF was the diluent and 25 μl of substrate stock solution was added to the cell suspension). After 0 to 60 min, 1.5 ml of the incubating cell suspension was removed, centrifuged at 14,000 rpm for 1 to 2 min in a Spectrafuge 16M Microcentrifuge (Labnet, Woodbridge, NJ, USA), and placed at −80 °C before HPLC injection.
The products of 4M5NC, 3A4M5NC, 4NBA, 4MC, and naphthalenediol were identified by comparing their HPLC retention times and UV visible spectra to standard chemicals and were corroborated through coelution (equal concentration of an authentic standard added to the newly identified product to verify its identity via the formation of a single-peak and absorbance spectra). To ensure the accuracy of the retention times, the HPLC column was equilibrated with the appropriate elution buffer before evaluation of various transformations. The authentic product standards 4M5NC, 3A4M5NC, 4NBA, 4MC, and naphthalenediol were also evaluated for their stability to determine appropriate sampling times. The negative control E. coli TG1/pBS(Kan) did not generate 4M5NC from 24DNT or naphthalenediol from naphthalene, and the negative control E. coli JVQ2/pBS(Kan) did not generate 3A4M5NC from 2A46DNT or 4NBA and 4MC from 4NT.
Initial product formation rates were determined by collecting supernatant samples at 2-, 5-, 10-, 15-, 20-, 30-, and 60-min incubation time points. Product formation rates were quantified in nanomole per minute per milligram of protein by converting product peak areas to concentrations using standard curves prepared at the specific absorbance wavelength for each product formed. The initial linear rates of product formation for the wild-type and engineered constructs were determined from at least two independent whole-cell transformation experiments. Protein content was 0.22 mg protein ml−1 OD−1 for recombinant E. coli TG1 and 0.18 mg protein ml−1 OD−1 for recombinant E. coli JVQ2 as determined using the Protein Assay Kit (Sigma Diagnostics). Authentic standards were not available for the products 2A46DNBA and 3A6M5NC; hence, for these products, relative product formation rates were determined using the initial linear plot of product peak area (at the maximum absorbance wavelength of each specific product) vs time and were normalized using the cell suspension OD and E. coli strain protein content.
Identification of 2A46DNBA, 3A4M5NC, and 3A6M5NC through gas chromatography–mass spectrometry
The formation of 2A46DNBA from 2A46DNT by wild-type DNTDDO and the formation of 3A6M5NC from 4A26DNT by wild-type NBDO was identified using gas chromatography–mass spectrometry (GC-MS) using a Hewlett-Packard 5970B GC-MS instrument equipped with a HP-1 column (12 m×0.2 mm, 0.33 μM thickness), and the ionization voltage was 70 eV. The initial column temperature was 120° C for 2 min and was increased at 10 to 270 °C/min followed by an isothermal operation for 6 min. The injector and detector temperatures were 270 and 275 °C. Whole-cell E. coli JVQ2/pBS(Kan)DNT and E. coli JVQ2/pBS(Kan)NBDO transformations were performed in a similar manner to the HPLC analyses, except that the cells were incubated for 1 h with 1 mM of 2A46DNT or 4A26DNT, respectively. After incubation for 1 h at 37 °C with each substrate, 20 ml of the incubating cell suspensions were mixed with 20 ml of ethyl acetate for 2 min, ultracentrifuged for 5 min at 6,000×g, the ethyl acetate layer was removed, and was evaporated under nitrogen gas. After the ethyl acetate was completely evaporated, the residue was dissolved in 40 μl of ethyl acetate and derivatized with 40 μl of N,O-bis(trimethylsilyl)-trifluoroacetamide (Alltech Associates, Deerfield, IL, USA), the samples were stored at −20 °C overnight before GS-MS injection.
Nitrite assay
The nitrite generated from the oxidation of all nitroaromatic compounds was detected spectrophotometrically as described previously (Keenan et al. 2004) based on the formation of an azo dye complex between nitrite, SUL, and N-(1-naphthyl)ethyldiamine (NAD); the azo dye has an maximum absorbance of 543 nm. The formations of 4M5NC from 24DNT, 3A4M5NC from 2A46DNT, 3A6M5NC from 4A26DNT, and 4MC from 4NT that were detected during the HPLC analyses were corroborated by the detection of nitrite (naphthalenediol did not interfere with nitrite detection).
For the 96-well nitrite assay, which was used to identify enzyme expression from correctly constructed plasmids, recombinant E. coli TG1 colonies were grown overnight at 37 °C, 250 rpm, in 270 μl of LB containing kanamycin (100 μg/ml) per well (96-well costar plate, Corning, Corning, NY, USA). After overnight growth (approximately 14 h) the 270 μl of cells was filtered using a 0.22-μm filter plate (Millipore, Billerica, MA, USA), washed once with 200 μl of 100 mM sodium phosphate buffer, pH 6.5, and resuspended with 200 μl of sodium phosphate buffer. Cells were incubated with 100 μM 24DNT, 250 μM 2A46DNT, or 250 μM 4A26DNT for 1 h at 37 °C. The formation of azo dye was evaluated at 540 nm (Labsystems Multiskan RC, Fisher Scientific, Pittsburgh, PA, USA) after the addition of 20 μl of SUL and NAD. The negative control, E. coli TG1 pBS(Kan), and the positive control, either E. coli TG1 pBS(Kan)R34 for 24DNT and 2A46DNT or E. coli TG1 pBS(Kan)NBDO for 4A26DNT, were each inoculated in triplicate per 96-well plate.
Results
Simultaneous degradation of 24DNT and naphthalene
To generate hybrid dioxygenases that can simultaneously degrade the aromatic pollutants 24DNT and naphthalene, the alpha- and beta-subunits (DntAc and DntAd, respectively) of R34DDO were added to the NDO dioxygenase (NagAaAbAcAd); hence, both terminal oxygenase components of NDO and R34DDO share a common ferredoxin reductase (NagAa) and ferredoxin (NagAb) (Fig. 2 and Fig. 3b). In the presence of naphthalene, the engineered orthric dioxygenase NDO-R34DDO increased the formation rate 4M5NC from 24DNT twofold relative to R34DDO alone (NDO does not oxidize 24DNT) (Table 3). In the presence of 24DNT, the formation rate of naphthalenediol from naphthalene by NDO-R34DDO was increased 68-fold relative to R34DDO alone (Table 3). Hence, the orthric dioxygenase was capable of the simultaneous oxidation of both substrates with wild-type-like rates of product formation, whereas neither NDO nor R34DDO alone had significant activity on 24DNT or naphthalene, respectively (Table 3).
Simultaneous degradation of 2A46DNT and 4A26DNT
Because TNT is easily reduced by biological systems (Yin et al. 2005), we generated hybrid dioxygenases that can simultaneously degrade the major TNT reduction products 2A46DNT and 4A26DNT; the alpha- and beta-subunits (NbzAc and NbzAd, respectively) of NBDO were added to R34DDO to form R34DDO-NBDO (Fig. 3c). As with NDO-R34DDO, in this novel dioxygenase construct, the two terminal oxygenases share a common electron transport system except here, the ferredoxin reductase (DntAa) and ferredoxin (DntAb) come from R34DDO rather than NDO.
GC-MS identified 2A46DNBA as the primary product generated from 2A46DNT by DNTDDO (Fig. 1); 2A46DNBA was also generated by R34DDO, NBDO, R34DDO-NBDO, NBDO-DNTDDO, and NDO-NBDO-DNTDDO. The product 2A46DNBA was identified with a GC-MS retention time of 12.1 min and the expected derivatized molecular ion [M+ (% relative intensity)] at m/z 285 (55); characteristic mass fragment ions were identified at m/z 270 (100), 194 (3), and 73 (68). To confirm the results of the HPLC and coelution analyses, 3A4M5NC was identified with GC-MS retention time of 11.0 min and the expected derivatized molecular ion [M+ (% relative intensity)] at m/z 328 (59); characteristic mass fragment ions were identified at 311 (44), 281 (66), 194 (19), and 73 (100). Also, GC-MS identified 3A6M5NC as the sole product generated by NBDO from 4A26DNT (Fig. 1); the 3A6M5NC product was also generated by R34DDO-NBDO and NDO-NBDO-DNTDDO. The product 3A6M5NC was identified with a GC-MS retention time of 11.8 min and the expected derivatized molecular ion [M+ (% relative intensity)] at m/z 328 (52); characteristic mass fragment ions were identified at m/z 311 (35), 281 (25), 194 (19), and 73 (100). The mass fragment ion results obtained through GC-MS identifications of 2A46DNBA, 3A4M5NC, and 3A6M5NC were similar to the results reported in previous studies (Johnson et al. 2001).
During the simultaneous degradation of 2A46DNT and 4A26DNT (0.25 mM each), R34DDO-NBDO generated 2A46DNBA threefold faster than NBDO, generated 3A4M5NC fourfold faster than NBDO, and generated 3A6M5NC infinitely faster than R34DDO (Table 4). Hence, the ability of R34DDO-NBDO to overcome the apparent inhibitory affect of simultaneous incubation with 2A46DNT and 4A26DNT exemplifies the utility of these novel constructs to enhance the degradation of the TNT reduction products.
To determine the ability of the orthric constructs to alter the product ratio of 2A46DNBA relative to 3A4M5NC, each compound was measured at its respective maximum absorbance of 300 and 350 nm after 60 min of incubation with 2A46DNT (data not shown). R34DDO generated 2A46DNBA relative to 3A4M5NC at a 6:1 ratio, DNTDDO at 3:1, NDO-R34DDO at 8:1, R34DDO-NBDO at 2:1, and NBDO-DNTDDO at 3:1. NBDO generated 2A46DNBA relative to 3A4M5NC at a ratio of 1:1 and NDO-NBDO-DNTDDO at 1:2; thus, these two enzymes favor the formation of 3A4M5NC. The NDO-NBDO-DNTDDO construct also increased the rate of 3A4M5NC twofold relative to NBDO without increasing the formation rate of 2A46DNBA (Table 4), showing the utility of this novel gene order to increase the formation rate of key intermediates in the degradation of nitroaromatic compounds.
Simultaneous degradation of 4A26DNT and naphthalene
To generate hybrid dioxygenases that can simultaneously degrade the TNT reduction product 4A26DNT and naphthalene, the alpha- and beta-subunits (NbzAc and NbzAd, respectively) of NBDO were added to NDO to form NDO-NBDO. In this novel dioxygenase construct, the two terminal oxygenases share a common ferredoxin reductase (NagAa) and ferredoxin (NagAb).
During the simultaneous degradation of 4A26DNT (0.25 mM) and naphthalene (0.1 mM), the construct NDO-NBDO generated naphthalenediol twofold faster than NBDO and generated 3A6M5NC infinitely faster than NDO because NDO has no activity on 4A26DNT (Table 4). The NDO-NBDO construct also generated naphthalenediol from naphthalene twofold faster than NBDO alone (Table 3).
Degradation of 4NT
Degradation of 4NT was also investigated using the constructs NDO-R34DDO and NDO-NBDO. The formation of 4NBA from 4NT by NDO-R34DDO was increased 5-fold relative to R34DDO and 1.5-fold relative to NDO (Table 4), showing that this novel dioxygenase has enhanced catalytic potential for a broad substrate range. Similarly, the formation rate of 4NBA from 4NT by NDO-NBDO was increased sevenfold relative to NBDO.
Protein expression analysis
SDS-PAGE on both E. coli TG1 and E. coli JVQ2 expressing pBS(Kan)R34, pBS(Kan)NDO−, pBS(Kan)NBDO, pBS(Kan)DNT, pBS(Kan)NDO-R34DDO, pBS(Kan)R34DDO-NBDO, pBS(Kan)NDO-NBDO, pBS(Kan)NBDO-DNTDDO, and pBS(Kan)NDO-NBDO-DNTDDO revealed that the expression of alpha- and beta-subunits in all these constructs was roughly equivalent.
Discussion
To date, this is the first report to prove the potential of a dual terminal oxygenase system for the simultaneous oxidation of environmental pollutants, and to have focused on the compatibility of the ferredoxin subunits and the nitroarene dioxygenase alpha-subunits. We show that the NagAb ferredoxin component from NDO is compatible with the terminal oxygenase alpha-subunits from both R34DDO and NBDO, and that the R34DDO DntAb ferredoxin component is compatible with the terminal oxygenase alpha-subunit of NBDO. We also show that inclusion of two oxygenase beta-subunits and two alpha-subunits in hybrid systems does not interfere with overall catalysis, and can lead to the increase in substrate ranges. The dioxygenase constructs, derived from altered gene order, were able to support wild-type-like rates of 4M5NC formation from 24DNT, naphthalenediol from naphthalene, 2A46DNBA/3A4M5NC from 2A46DNT, 3A6M5NC from 4A26DNT, and 4MC/4NBA from 4NT. This is the first report to describe a dioxygenase system capable of oxidizing mixtures of 24DNT and naphthalene via the enzymes NDO and R34DDO. The substrate range of the novel NDO-R34DDO construct is significant considering that attempts to convert a NDO into a dioxygenase capable of oxidizing 24DNT through protein engineering were unsuccessful (Keenan et al. 2005; Parales et al. 2000b), and the enhancement of R34DDO activity toward naphthalene through saturation mutagenesis led to loss of activity of R34DDO to degrade 24DNT (Keenan et al. 2004).
This is also the first report to describe a dioxygenase system capable of oxidizing the mixtures of 2A46DNT and 4A26DNT via the enzymes R34DDO, NBDO, and NDO-NBDO-DNTDDO. The enhancement in the formation of 3A4M5NC over 2A46DNBA during the oxidation of 2A46DNT by NDO-NBDO-DNTDDO clearly shows the usefulness of this novel construct for generating the initial intermediate needed for the potential oxidative degradation pathway of 2A46DNT. The requirement of catechols rather than benzyl alcohols during oxidative mineralization of nitroaromatics is intuitive because the utilization of 24DNT as a growth substrate by both B. cepacia R34 and Burkholderia sp. strain DNT and nitrobenzene by Comamonas sp. strain JS765 requires the formation of dihydroxylated catechols rather than benzyl alcohol (Nishino et al. 2000; Nishino and Spain 1995; Spanggord et al. 1991).
Though only mixtures of 24DNT/naphthalene, 2A46DNT/4A26DNT, and 4A26DNT/naphthalene were evaluated in this study, the results presented here suggest that the NDO-R34DDO, R34DDO-NBDO, NDO-NBDO-DNTDDO, and NDO-NBDO orthric dioxygenases would be able to simultaneously degrade mixtures of 24DNT, naphthalene, 4NT, 2A46DNT, and 4A26DNT. The substrates evaluated in this study also represent a small fraction of the total number of substrates, and therefore, substrate combinations that can potentially be oxidized by the enzymes of these orthric constructs (Keenan et al. 2004, 2005; Lessner et al. 2002; Leungsakul et al. 2005; Nishino et al. 2000). For example, NDO-R34DDO should be able to simultaneously degrade other compounds such as phenanthrene (degraded by NDO (Fuenmayor and Lemoine 1992)) and 26DNT (degraded by R34DDO; Nishino et al. 2000). The ability of NDO-NBDO to degrade structurally-diverse substrates, such as 4A26DNT and naphthalene, one substituted aromatic compound, and one polycyclic aromatic compound, shows the usefulness of this construct in degrading complex mixtures of pollutants.
From the high enzymatic activities reported here it can be inferred that the amino acid similarity of the ferredoxin component is more important than the amino acid similarity of the terminal oxygenase alpha-subunit for dictating the performance of a hybrid Rieske dioxygenase systems. This is shown by considering that R34DDO DntAc, DNTDDO DntAc, and NBDO NbzAc share 20.5, 20.6, and 21.2% amino acid identity with NagG, respectively; yet, all four terminal oxygenase components can utilize the same NDO NagAb ferredoxin component. The promiscuity of the ferredoxin subunit of the NDO (NahAb) from P. putida G7 and the BPDO (BphF) from Comamonas testosteroni B-356 was reported (Barriault and Sylvestre 1999); however, the activity of the swapped dioxygenases were 5- to 50-fold less active toward biphenyl than the wild-type dioxygenases, showing that the ferredoxin subunits may not be interchanged for developing dioxygenase systems with enhanced degradation capabilities. This indicates that the hybrid system should contain the ferredoxin and ferredoxin reductase from the same system. Recently, it was reported that the ferredoxin component of NCIB NDO could substitute for the ferredoxin component from 2NTDO during the oxidation of 2NT; however, the ferredoxin component from BPDO or the toluene dioxygenase from P. putida F1 were not compatible with the 2NTDO terminal oxygenase (Parales et al. 2005). This supports the research presented in our report, and our research provides results to show that the ferredoxin reductase and ferredoxin components from nitroarene and NDO systems can support two different terminal oxygenases for the degradation of mixtures 24DNT/naphthalene, 2A46DNT/4A26DNT, and 4A26DNT/naphthalene.
This report introduces a novel gene order that was not previously reported for a naphthalene Rieske nonheme iron dioxygenase system. For the constructs presented here, a terminal dioxygenase is inserted after the ferredoxin reductase gene and before the ferredoxin gene (Table 1) for NDO-R34DDO, R34DDO-NBDO, NDO-NBDO, NBDO-DNTDDO, and NDO-NBDO-DNTDDO. For the first time, hybrid gene constructs from the NDO and the nitroarene dioxygenases (R34DDO, DNTDDO, and NBDO) showed these loci tolerate genetic engineering and rearrangement. This tolerance represents a scientific finding that may have been conceptually expected, but the experimental evidence of this capability presented here may lead to further optimization with regard to transcript stability, catalytic activity, and future multiple terminal oxygenase constructs. The incorporation of mutant alpha-subunits into various orthric constructs may further increase the application of Rieske dioxygenase systems for green chemistry and bioremediation applications. The order of the terminal oxygenase genes (bphA1A2 and todC1C2) before the ferredoxin reductase gene (bphA3 and todA) and ferredoxin gene (bphA4 and todB) for the wild-type biphenyl (bphA1A2A3A4) (Taira et al. 1992) and toluene dioxygenase systems (todC1C2AB) (Zylstra and Gibson 1989) are different from the wild-type gene order for NDO, NBDO, R34DDO, and DNTDDO. It should be noted that the ferredoxin gene is encoded after the beta-subunit in the engineered constructs reported here and also in the wild-type bphA and todC operons. The only wild-type example of the dioxygenase gene order presented here exists in the BPDO gene cluster from C. testosteroni B-356; however, in this strain the ferredoxin reductase gene (bphG) is separated from the alpha-subunit gene (bphA) by over 15 kb (Sylvestre et al. 1996).
For the construct NDO-NBDO-DNTDDO, the differences in the amino acid sequence between DntAc and NbzAc and therefore the alpha-subunit three-dimensional confirmation may favor the association of the NBDO alpha-subunit with the NDO ferredoxin component (NagAb) over the association of DntAc with NagAb. This is exemplified by the activity of NDO-NBDO vs that of NDO-NBDO-DNTDDO for the formation of 3A6M5NC from 4A26DNT where NbzAc could out-compete DntAc for association with NagAb (showing an increase in the formation rate of 3A6M5NC, Table 4), but could not out-compete NagAc for association with NagAb in the NDO-NBDO construct (showing a decrease in the formation rate of 3A6M5NC, Table 4). It will be valuable to investigate whether the random mutagenesis of a ferredoxin gene will cause increases in substrate oxidation rates by allowing the ferredoxin component to become more compatible with two different terminal oxygenase alpha-subunits.
While the crystal structure of the ferredoxin subunit was solved for BphF of B. cepacia LB400 (Colbert et al. 2000) and CarAc of P. resinovorans strain CA10 (Nam et al. 2005), the factors that determine the assembly of the active Rieske dioxygenase are not known. Both reports which have described the structure of the Rieske ferredoxin component clearly express that the topology and surface charge, which are dictated by the ferredoxin amino acid sequence, govern the interactions with the alpha-subunit of the terminal oxygenase (Colbert et al. 2000; Nam et al. 2005). These novel dual terminal oxygenase constructs may ultimately aid in determining the importance of specific amino acids that govern the association of the ferredoxin component with the terminal oxygenase of Rieske dioxygenase systems. In addition, this engineering technique is general and may be applied to other dioxygenase systems and to other multisubunit proteins like toluene monooxygenases or may be used to combine an engineered monooxygenase and a dioxygenase (similar to the wild-type Ralstonia sp. strain U2 system). One can envision combining a myriad of enzymes to create hybrid enzymes with even more diverse activities because many biological catalysts rely on NADH.
References
ATSDR (2004) Public Health Assessment Volunteer Army Ammunition Plant Chattanooga, Hamilton County, Tennessee. Agency for Toxic Substances and Disease Registry, Atlanta, GA
Barriault D, Sylvestre M (1999) Functionality of biphenyl 2,3-dioxygenase components in naphthalene 1,2-dioxygenase. Appl Microbiol Biotechnol 51:592–597
Canada KA, Iwashita S, Shim H, Wood TK (2002) Directed evolution of toluene ortho-monooxygenase for enhanced 1-naphthol synthesis and chlorinated ethene degradation. J Bacteriol 184:344–349
Colbert CL, Couture MM-J, Eltis LD, Bolin JT (2000) A cluster exposed: structure of the Rieske ferredoxin from biphenyl dioxygenase and the redox properties of Rieske Fe-S proteins. Structure 8:1267–1278
Erickson BD, Mondello FJ (1993) Enhanced biodegradation of polychlorinated biphenyls after site-directed mutagenesis of a biphenyl dioxygenase gene. Appl Environ Microbiol 59:3858–3862
Fuenmayor SL, Lemoine VR (1992) Characterization of polycyclic aromatic hydrocarbons degradative soil Pseudomonas. Acta Cient Venez 43:349–354
Fuenmayor SL, Wild M, Boyes AL, Williams PA (1998) A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J Bacteriol 180:2522–2530
Gantz T (1993) Early Greek myth: a guide to literary and artistic sources. The Johns Hopkins University Press, Baltimore, pp 133–134
Green RD (1998) Five year review: Wamchem superfund site Beaufort, South Carolina. Environmental Protection Agency, Atlanta, Georgia, pp 1–15
Howe-Grant M (1991) In: Howe-Grant M (ed) Kirk–Othmer encyclopedia of chemical technology. Wiley-Interscience Publishers, New York, pp 133–152
Johnson GR, Smets BF, Spain JC (2001) Oxidative transformation of aminodinitrotoluene isomers by multicomponent dioxygenases. Appl Environ Microbiol 67:5460–5466
Johnson GR, Jain RK, Spain JC (2002) Origins of the 2,4-dinitrotoluene pathway. J Bacteriol 184:4219–4232
Johnson LR, Davenport R, Balbach H, Schaeffer DJ (1994) Phototoxicity 3. Comparative toxicity of trinitrotoluene and aminodinitrotoluenes to Daphnia magna, Dugesia dorotocephala, and sheep erythrocytes. Ecotoxicol Environ Saf 27:34–49
Keenan BG, Leungsakul T, Smets BF, Wood TK (2004) Saturation mutagenesis of Burkholderia cepacia R34 2,4-dinitrotoluene dioxygenase at DntAc valine 350 for synthesizing nitrohydroquinone, methylhydroquinone, and methoxyhydroquinone. Appl Environ Microbiol 70:3221–3222
Keenan BG, Leungsakul T, Smets BF, Mori M, Henderson DE, Wood TK (2005) Protein engineering of the archetypal nitroarene dioxygenase of Ralstonia sp. strain U2 for activity on aminonitrotoluenes and dinitrotoluenes through alpha-subunit residues leucine 225, phenylalanine 350, and glycine 407. J Bacteriol 187:3302–3310
Keith LH, Telliard WA (1979) Priority pollutants. Environ Sci Technol 13:416–423
Kumamaru T, Suenaga H, Mitsuoka M, Watanabe T, Furukawa K (1998) Enhanced degradation of polychlorinated biphenyls by directed evolution of biphenyl dioxygenase. Nat Biotechnol 16:663–666
Lee K (1998) Involvement of electrostatic interactions between the components of toluene dioxygenase from Pseudomonas putida F1. J Microbiol Biotechnol 8:416–421
Lessner DJ, Johnson GR, Parales RE, Spain JC, Gibson DT (2002) Molecular characterization and substrate specificity of nitrobenzene dioxygenase from Comamonas sp. strain JS765. Appl Environ Microbiol 68:634–641
Leungsakul T, Keenan BG, Yin H, Smets BF, Wood TK (2005) Saturation mutagenesis of 2,4-DNT dioxygenase of Burkholderia sp. strain DNT for enhanced dinitrotoluene degradation. Biotechnol Bioeng 92:510–517
Mori M, Shoji M, Sayama M, Kondo T, Inoue M, Kodaira K (2000) Secondary metabolism of dinitrobenzyl glucuronide related to production of genotoxic compounds of dinitrotoluene in male Wistar rat. J Health Sci 46:329–335
Nam J-W, Noguchi H, Fujimoto Z, Mizuno H, Ashikawa Y, Abo M, Fushinobu S, Kobashi N, Wakagi T, Iwata K et al (2005) Crystal structure of the ferredoxin component of carbazole 1,9a-dioxygenase of Pseudomonas resinovorans strain CA10, a novel Rieske non-heme iron oxygenase system. Proteins 58:779–789
Nishino SF, Spain JC (1995) Oxidative pathway for the biodegradation of nitrobenzene by Comamonas sp. strain JS765. Appl Environ Microbiol 61:2308–2313
Nishino SF, Paoli GC, Spain JC (2000) Aerobic degradation of dinitrotoluenes and pathway for bacterial degradation of 2,6-dinitrotoluene. Appl Environ Microbiol 66:2139–2147
Parales JV, Parales RE, Resnick SM, Gibson DT (1998a) Enzyme specificity of 2-nitrotoluene 2,3-dioxygenase from Pseudomonas sp. strain JS42 is determined by the C-terminal region of the alpha-subunit of the oxygenase component. J Bacteriol 180:1194–1199
Parales RE, Emig MD, Lynch NA, Gibson DT (1998b) Substrate specificities of hybrid naphthalene and 2,4-dinitrotoluene dioxygenase enzyme systems. J Bacteriol 180:2337–2344
Parales RE, Lee K, Resnick SM, Jiang H, Lessner DJ, Gibson DT (2000a) Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme. J Bacteriol 182:1641–1649
Parales RE, Resnick SM, Yu C-L, Boyd DR, Sharma ND, Gibson DT (2000b) Regioselectivity and enantioselectivity of naphthalene dioxygenase during arene cis-dihydroxylation: control by phenylalanine 352 in the alpha-subunit. J Bacteriol 182:5495–5504
Parales RE, Huang R, Yu C-L, Parales JV, Lee FKN, Lessner DJ, Ivkovic-Jensen MM, Liu W, Friemann R, Ramaswamy S and others (2005) Purification, characterization, and crystallization of the components of the nitrobenzene and 2-nitrotoluene dioxygenase enzyme systems. Appl Environ Microbiol 71:3806–3814
Rui L, Reardon K, Wood TK (2005) Protein engineering of toluene ortho-monooxygenase of Burkholderia cepacia G4 for regiospecific hydroxylation of indole to form various indigoid compounds. Appl Microbiol Biotechnol 66:422–429
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Spanggord RJ, Spain JC, Nishino SF, Mortelmans KE (1991) Biodegradation of 2,4-dinitrotoluene by a Pseudomonas sp. Appl Environ Microbiol 57:3200–3205
Stohs SJ, Ohia S, Bagchi D (2002) Naphthalene toxicity and antioxidant nutrients. Toxicology 180:97–105
Suen WC, Haigler BE, Spain JC (1996) 2,4-Dinitrotoluene dioxygenase from Burkholderia sp. strain DNT: similarity to naphthalene dioxygenase. J Bacteriol 178:4926–4934
Sylvestre M, Sirois M, Hurtubise Y, Bergeron J, Ahmad D, Shareck F, Barriault D, Guillemette I, Juteau JM (1996) Sequencing of Comamonas testosteroni strain B-356-biphenyl/chlorobiphenyl dioxygenase genes: evolutionary relationships among gram-negative bacterial biphenyl dioxygenases. Gene 174:195–202
Taira K, Hirose J, Hayashida S, Furukawa K (1992) Analysis of bph operon from the polychlorinated biphenyl-degrading strain of KF707. J Biol Chem 267:4844–4853
Whiteway J, Koziarz P, Veall J, Sandhu N, Kumar P, Hoecher B, Lambert IB (1998) Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J Bacteriol 180:5529–5539
Yin H, Wood TK, Smets BF (2005) Reductive transformation of TNT by Escherichia coli: pathway description. Appl Microbiol Biotechnol 67:397–404
Zylstra GJ, Gibson DT (1989) Toluene degradation by Pseudomonas putida F1. J Biol Chem 264:14940–14946
Acknowledgements
This study was supported by the National Science Foundation (BES-0114126). We thank P. A. Williams for plasmid pWWF6, I. B. Lambert for E. coli JVQ2, J. C. Spain for 4M5NC, 3A4M5NC, and for plasmid pJS765, A. Fishman for her assistance with the HPLC analyses, and M. Thompson for his assistance with the GC-MS analyses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Keenan, B.G., Wood, T.K. Orthric Rieske dioxygenases for degrading mixtures of 2,4-dinitrotoluene/naphthalene and 2-amino-4,6-dinitrotoluene/4-amino-2,6-dinitrotoluene. Appl Microbiol Biotechnol 73, 827–838 (2006). https://doi.org/10.1007/s00253-006-0538-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0538-8