
Abstract
cDNAs encoding two glycoside hydrolase family 1 β-glucosidases (BGL1A and BGL1B) were cloned from the basidiomycete Phanerochaete chrysosporium, and the substrate specificities of the recombinant enzymes and the expression patterns of the two genes were investigated in relation to cellobiose metabolism by the fungus. The cDNA sequences contained open reading frames of 1,389 base pairs (bp) (bgl1A) and 1,623 bp (bgl1B), encoding 462 and 530 amino acids, respectively. Although high sequence identity (65%) was observed between the deduced amino acid sequences of the two enzymes, an apparent difference was observed at the C-terminal region: BGL1B has a 63-amino acid extension, which has no similarity with any known protein. Both recombinant enzymes expressed in Escherichia coli showed hydrolytic activity towards several β-glycosidic compounds. However, the substrate recognition patterns of the two enzymes were quite different from each other. In particular, cellobiose was hydrolyzed more effectively by BGL1B than by BGL1A. The expression of the two genes in the fungus was monitored by reverse transcription-PCR, which showed that bgl1A was expressed constitutively in both glucose- and cellobiose-containing culture, whereas bgl1B was expressed in cellobiose culture but was repressed in glucose culture, possibly because of carbon catabolite repression. We conclude that BGL1B contributes to cellobiose metabolism during cellulose degradation by P. chrysosporium.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In filamentous fungi, the biodegradation of cellulose has been considered to proceed by two-step hydrolysis, i.e., cellulose is first degraded to cellobiose by cellobiohydrolases and endo-glucanases and then cellobiose is hydrolyzed to glucose by extracellular β-glucosidases (BGLs). However, Eriksson and coworkers have demonstrated the importance of oxidative enzymes in cellulose degradation, and have isolated cellobiose dehydrogenase (CDH) from the cellulolytic culture of the wood-rotting basidiomycete Phanerochaete chrysosporium (Ayers et al. 1978; Eriksson et al. 1974; Westermark and Eriksson 1974). Because this enzyme effectively oxidizes cellobiose to cellobionolactone, two independent extracellular pathways for cellobiose metabolism, hydrolysis by BGL and oxidation by CDH, have been proposed to be involved in cellulose degradation by the fungus (Eriksson 1978). In our previous studies, however, we demonstrated that the fungus produces only one extracellular BGL, and the hydrolysis of cellobiose by the enzyme is considerably less active than the oxidation of cellobiose by CDH (Igarashi et al. 2003; Yoshida et al. 2001). Moreover, when the substrate specificity of the extracellular BGL was compared among several glucosides, β-1,3-glucan was the best substrate. In addition, transcription of the BGL gene was repressed in the presence of cellobiose, whereas it was induced in the absence of any carbon source (Yoshida et al. 2004). These results suggest that the extracellular BGL, which was believed to hydrolyze cellobiose, does not participate in cellobiose metabolism during extracellular cellulose degradation, but rather plays a role in reutilization of the fungal cell wall.
Glycoside hydrolases (GHs) are now classified into more than 100 families, whereas BGLs from filamentous fungi are categorized into only two GH families, families 1 and 3, according to the Carbohydrate-Active enZymes server (CAZy, http://www.cazy.org/CAZY/) (Henrissat 1991; Henrissat and Bairoch 1993, 1996). The cDNA encoding the extracellular BGL from P. chrysosporium described above has been cloned, and the deduced amino acid sequence suggested that the enzyme can be classified into GH family 3 (Kawai et al. 2003; Li and Renganathan 1998; Lymar et al. 1995). Recently, the total genome sequence of P. chrysosporium has been disclosed (http://genome.jgi-psf.org/Phchr1/Phchr1.home.html), and it has been suggested that the fungus has two putative BGLs belonging to GH family 1 (Martinez et al. 2004). The occurrence of GH family 1 BGLs as intracellular enzymes has already been reported in cellulolytic ascomycetes such as Trichoderma (Hypocrea) spp. (Saloheimo et al. 2002; Takashima et al. 1999), Humicola spp. (Peralta et al. 1990; Takashima et al. 1996, 1999), and Talaromyces spp. (Mchale and Coughlan 1981a,b), suggesting that the basidiomycete P. chrysosporium utilizes similar enzymes for cellobiose metabolism. As far as we know, however, there is no report about GH family 1 BGL from basidiomycetes.
In the present work, we have cloned cDNAs encoding two GH family 1 BGLs from the basidiomycete P. chrysosporium and successfully produced the recombinant enzymes in an Escherichia coli expression system. The kinetic features of the two recombinant enzymes for cellobiose and the transcription of their genes were investigated in relation to cellobiose metabolism.
Materials and methods
Cloning of cDNAs encoding GH family 1 BGLs
Phanerochaete chrysosporium K-3 (Johnsrud and Eriksson 1985) was grown on the Kremer and Wood medium (Kremer and Wood 1992) containing 2% cellulose (CF11; Whatman, Clifton, NJ, USA), 0.2% cellobiose, or 0.2% glucose as a sole carbon source, based on a previous report (Habu et al. 1997). After 3 days of cultivation, mycelia were collected by filtration and frozen with liquid nitrogen. Total RNA was extracted from the mycelial powder, and first-strand cDNA was synthesized as described previously (Kawai et al. 2005). The oligonucleotide primers were designed based on P. chrysosporium genome information, as illustrated in Fig. 1.

Schematic diagram of cDNA encoding P. chrysosporium BGL1A (a) and BGL1B (b), showing the locations and nucleotide sequences of primers used for PCR. F and R indicate the forward and reverse directions, respectively
Transcriptional analysis
The first-strand cDNA from each sample was used for reverse transcription (RT) PCR analysis with bgl1A-F 5′-ACCGATGCAAGCAAGGA-3′ and bgl1A-R 5′-TCGAAGTTATCGAGAAGACTCCAT-3′ for bgl1A gene and bgl1B-F 5′-GATGTTGCCATCGGTTGG-3′ and bgl1B-R 5′-AAGTTGTCGAGAAGACTCCAGC-3′ for bgl1B gene. The transcriptional level of the actin gene (accession number AB115328), a housekeeping gene, was measured using primers actin-F 5′-GCCGTGTTCCCGTCCAT-3′ and actin-R 5′-CACTTGTAGATGGAGTTGTACGTCGT-3′.
Sequence analysis
A database search using the deduced amino acid sequences was performed using blastp (http://www.ncbi.nlm.nih.gov/BLAST/) (Altschul et al. 1990, 1997). Comparative analysis of homology with BGL1A and BGL1B was performed using CLUSTAL W (http://www.ebi.ac.uk/clustalw/) (Thompson et al. 1994) with default parameters. To predict the localizations of the two enzymes, the amino acid sequences of the two enzymes were scanned for the presence of signal peptides using the SignalP version 3.0 (Bendtsen et al. 2004; Nielsen et al. 1997) server at the Center for Biological Sequence Analysis (http://www.cbs.dtu.dk/services/SignalP/).
Production of recombinant BGLs
The oligonucleotide primers BGL1A-F 5′-TTTCCATGGCGATGTCTGCCGCGAAACTACCCAA-3′ and BGL1A-R 5′-TTTAAGCTTTCAATGATGATGATGATGATGCTCTTCGATGTGCTCCTT-3′, BGL1B-F 5′-TTTCCATGGCGATGTCTGCGTCCGCCGCTCCT-3′, and BGL1B-R 5′-TTTAAGCTTTCATGATGATGATGATGATGGAGGCCGAGGAATGCAGA-3′, introducing NcoI and HindIII cleavage sites (in bold face) and His6-tag (underlined sequence), were used to amplify the BGL1A and BGL1B coding sequences. Both genes were cloned into the expression vector pET 28a (+) (Novagen, Madison, WI, USA), and the vectors pET-BGL1A and pET-BGL1B thus obtained were transformed into the E. coli host strain Rosetta (DE3) (Novagen). The kanamycin-resistant transformants were grown overnight in 200 ml of Luria-Bertani (LB) medium supplemented with 20 μg/ml kanamycin at 37°C. The culture (10 ml) was inoculated into 1 l of same medium and the culture was grown at 37°C until the OD600 reached 0.5–0.6. Then, isopropylthiogalactoside was added to a final concentration of 50 μM and the cultures were grown at 26.5°C for 4 h. Cells were harvested by centrifugation at 3,000×g for 10 min, and stored at −80°C.
Purification of recombinant BGLs
Induced cells (500 ml culture) were resuspended in 50 ml of 20 mM potassium phosphate buffer (pH 7.0) supplemented with 1 mM EDTA and 1 mM phenylmethanesulfonyl fluoride and disrupted by sonication in a Bioruptor UCD-200T (COSMO BIO, Tokyo, Japan). All subsequent manipulations were carried out at 4°C, except for the enzyme assay. The cell debris was eliminated by centrifugation at 10,000×g for 30 min. The supernatant was concentrated by adding ammonium sulfate to 70% final saturation, then the precipitate was dissolved in 50 ml of 20 mM methanesulfonic acid (MES) (pH 6.0) with 1 M ammonium sulfate. The resulting solution was applied to a Phenyl-Toyopearl 650S (Tosoh, Tokyo, Japan) column (22×150 mm) equilibrated with 20 mM MES (pH 6.0) containing 1 M ammonium sulfate and eluted with a linear reverse gradient of ammonium sulfate from 1.0 to 0 M in 100 ml. Fractions were assayed for p-nitrophenyl-β-d-glucoside (pNP-Glu, Sigma-Aldrich, St. Louis, MO, USA) hydrolyzing activity, as described in the following “Enzyme assays” section. The active fractions were pooled and concentrated in a stirred ultrafiltration cell (Model 8050, Millipore, Billerica, MA, USA) equipped with an ultrafiltration disc (Biomax-10, Millipore). The enzyme solution was applied to a diethylaminoethyl-Toyopearl 650S (Tosoh) column (16×150 mm) equilibrated with 20 mM Tris–HCl (pH 8.0) and eluted with a linear gradient of potassium chloride from 0 to 0.3 M in 240 ml. The active fractions were pooled, concentrated, applied to a Ni-Sepharose high-performance (GE Healthcare Bio-Sciences, Piscataway, NJ, USA) column (16×80 mm) equilibrated with 20 mM 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (pH 7.4) containing 0.5 M sodium chloride, and eluted with a linear gradient of imidazole from 0 to 0.3 M in 50 ml. Fractions containing BGL activity were pooled and concentrated by centrifugal filtration (Vivaspin 15, Sartorius AG, Goettingen, Germany). The enzyme was judged to be homogeneous by sodium dodecyl sulfate polyacrylamide gel electrophoresis [12% (w/v) polyacrylamide] and the concentrations of the two BGLs were determined from the absorbance at 280 nm (BGL1A; ɛ 280=123 cm−1mM−1, BGL1B; ɛ 280=126 cm−1mM−1).
Enzyme assays
p-Nitrophenyl-β-d-galactoside (pNP-Gal), p-nitrophenyl-β-d-xyloside (pNP-Xyl), and p-nitrophenyl-β-d-mannoside (pNP-Man) were purchased from Sigma-Aldrich. Enzyme activities towards pNP-Glu, pNP-Gal, pNP-Xyl, and pNP-Man were determined by monitoring the amount of released p-nitrophenol at various substrate concentrations in 50 mM MES buffer, pH 6.5, at 30°C, as described previously (Igarashi et al. 1997). Activities of BGL toward cellobiose (Seikagaku, Tokyo, Japan) were determined by monitoring the amount of released glucose at various substrate concentrations. The enzymatic reactions were stopped by boiling the reaction mixture for 3 min. The amount of glucose released by BGL activity was detected using Glucose CII-Test Wako (Wako Pure Chemical, Osaka, Japan) after 10 min incubation in 50 mM MES buffer, pH 6.5, at 30°C, as described previously (Kawai et al. 2003). The obtained data were fitted to the Michaelis–Menten equation. To calculate the kinetic parameters for substrate transglycosylation, in the case of cellobiose, the following equation was used for the curve fitting (Kawai et al. 2004):
Where K m is the Michaelis constant, K m2 is the transglycosylation constant, k cat is the catalytic constant, and k cat2 is the maximum transglycosylation activity. All kinetic parameters were calculated using Delta Graph 5.5.5 (Red Rock, Salt Lake City, UT, USA) and KaleidaGraph 3.6.4 (Synergy, Reading, PA, USA).
Nucleotide sequence accession number
The nucleotide sequences of the cDNAs encoding P. chrysosporium BGL1A and BGL1B have been submitted to the DNA Data Bank of Japan/European Molecular Biology Laboratory/GenBank databases under accession numbers AB253326 and AB253327, respectively.
Results
Cloning of two cDNAs encoding GH family 1 BGLs
Based on the nucleotide sequences named gx.44.14.1 and gx.79.11.1 in the total genomic sequences of P. chrysosporium, primers were designed as shown in Fig. 1, and two cDNAs encoding putative GH family 1 BGLs were cloned by RT-PCR. The gene in gx.44.14.1, termed bgl1A, contained an open reading frame (ORF) consisting of 1,389 base pairs (bp) encoding 462 amino acid residues (ESM Appendix 1). The gene in gx.79.11.1, termed bgl1B, contained an ORF consisting of 1,623 bp coding 540 amino acid residues (ESM Appendix 1).
BGL1A and BGL1B showed high sequence identities (more than 50%) with other GH family 1 BGLs from the ascomycetes. BGL1A and BGL1B had the highest identities with bgl4 from Humicola grisea, 56 and 58%, respectively. Two glutamic acids corresponding to the putative catalytic proton donor and nucleophile, Glu170 and Glu365 (BGL1A) and Glu175 and Glu380 (BGL1B), are conserved in P. chrysosporium BGLs, as well as in other fungal BGLs belonging to GH family 1 (Fig. 2). When the amino acid sequences of BGL1A and BGL1B were compared with each other, BGL1B was found to have a 63-amino acid extension at the C-terminal region, which does not show similarity with any known protein in the BLAST search. Excluding the extension, 65% identity was obtained for the catalytic domain, where the catalytic residues are conserved, suggesting BGL1A and BGL1B may have similar structural features. To predict the localization of these enzymes, web-based analysis of their amino acid sequences with SignalP was performed. The program predicted that both BGL1A and BGL1B are intracellular proteins because there is no secretion signal at the N-terminal region.

Multiple alignment of amino acid sequences of P. chrysosporium BGL1A, BGL1B, and other BGLs from ascomycetes belonging to GH family 1. The conserved GH family 1 N-terminal signature is highlighted. The putative catalytic proton donor is boxed, and the putative catalytic nucleophile is marked with half-toned boxes. bgl4, H. grisea var. thermoidea bgl4 (AB003109); Bgl2, Hypocrea jecorina QM9414 Bgl2 (AB003110); Bg1, Talaromyces emersonii Bg1 (AF439322)
Substrate specificities of two recombinant BGLs
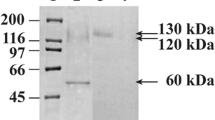
The bgl1A and bgl1B genes were expressed with a C-terminal His6-tag in E. coli and purified by three-step column chromatography. The purified BGL1A and BGL1B each gave a single band with molecular weights of 53 and 60 kDa, respectively, as shown in Fig. 3. The recombinant enzymes successively hydrolyzed several glycosidic substrates, indicating that the recombinant proteins are correctly folded. The substrate specificities of the two BGLs are summarized in Table 1. In both cases, pNP-Glu was hydrolyzed more effectively than other substrates, although apparent activity was detected when pNP-Gal was used as the substrate. Little or no activity was seen with pNP-Xyl and pNP-Man, suggesting that recognition at the C2 hydroxyl group and C6 methylol group is strict in the active site of the enzymes.

Sodium dodecyl sulfate polyacrylamide gel electrophoresis of purified recombinant BGL1A and BGL1B. S, molecular weight standards (kDa); lane 1, BGL1A; Lane 2, BGL1B
To evaluate cellobiose hydrolysis, the substrate concentration dependence of hydrolytic velocity by BGL1A and BGL1B was compared with that of extracellular BGL3A, as shown in Fig. 4. With the increase of substrate concentration above 1 mM cellobiose, the specific activity of BGL1B decreased. This is presumably because of transglycosylation, as reported in the case of BGL3A (Kawai et al. 2004) because an additional triose spot was observed by thin-layer chromatography in the reaction mixture of BGL1B (data not shown). Although the K m values of BGL1A (6.80±0.29 mM) and BGL3A (3.35±0.32 mM) (Igarashi et al. 2003) are in the same range, the affinity of BGL1B for cellobiose was quite high, with a K m of 0.218±0.017 mM, indicating a kinetic advantage of BGL1B over other BGLs.

Hydrolysis of cellobiose by BGLs from P. chrysosporium. Hydrolytic velocity for cellobiose was estimated from the amount of glucose released by each enzyme, as described in the “Materials and methods” section. Filled squares, BGL1A; filled circles, BGL1B; open squares, BGL3A. The plot of BGL3A is taken from a previous report (Igarashi et al. 2003)
Transcriptional analysis of P. chrysosporium BGL1s
To monitor the gene expression of bgl1A and bgl1B in glucose and cellobiose culture, the amounts of their transcripts were monitored by RT-PCR. Because the primers had been designed not to include intron regions, the obtained results could be directly compared without interference from contaminating genomic DNA. As shown in Fig. 5, a significant difference was observed in the transcription of BGL1B between glucose and cellobiose cultures, whereas BGL1A was expressed equally in both cultures, compared with the expression of actin gene as a house-keeping gene. This suggests that the transcriptional response of bgl1B is similar to that of other cellulose-degrading enzymes with carbon catabolite repression (Broda et al. 1995; Tempelaars et al. 1994).

RT-PCR analysis of the expression of bgl1A (a), bgl1B (b), and actin (c) genes in mycelia grown on glucose and cellobiose-containing media. Lane 1, glucose culture; lane 2, cellobiose culture. The PCR products were separated on a 2% agarose gel
Discussion
Cellobiose metabolism is one of the important steps of cellulose degradation, and extracellular BGL has been thought to act as a cellobiase in filamentous fungi. However, previous studies demonstrated that P. chrysosporium produces only one extracellular BGL belonging to GH family 3 (BGL3A), and the enzyme seems unlikely to be involved in cellobiose metabolism from both kinetic and transcriptional points of view. To find the enzyme responsible for cellobiose metabolism, therefore, we have focused in the present study on the kinetic features of GH family 1 BGLs and the expression patterns of the genes.
There are numerous reports about GH family 1 BGLs from ascomycetes Trichoderma, Humicola, and Talalomyces species, although information about basidiomycetes BGL1 is limited. In ascomycetes, all enzymes are characterized as intracellular BGLs because they are purified from the mycelium, but not from the culture solution. The sequence analysis in the present study indicates that P. chrysosporium BGL1A and BGL1B both lack a secretion signal, suggesting they operate intracellularly. Both recombinant P. chrysosporium BGLs have hydrolytic activity towards various glycosidic compounds. Similar substrate recognition patterns for pNP-glycosidic substrates indicate that BGL1A and BGL1B have similar environments in the subsite −1. In contrast, the kinetic parameters of the two enzymes for cellobiose hydrolysis were quite different. This might be because of a difference at subsite +1. In addition, substrate transglycosylation has been observed in BGL1B with a quite low K m2 value (3.20±0.69 mM), but not in BGL1A; this again suggests a difference in affinity for a glucose unit at the subsite +1. The kinetic efficiency (k cat/K m) of BGL1B (75 s−1mM−1) for cellobiose was 280 and 14 times higher than those of BGL1A (0.27 s−1mM−1) and BGL3A (5.5 s−1mM−1), respectively. This result clearly shows a kinetic advantage of BGL1B for cellobiose metabolism, compared with BGL1A or BGL3A.
Although transcription of bgl1A was detected in glucose culture as well as cellobiose culture, that of bgl1B was apparently repressed in glucose culture, whereas a significant expression level of bgl1B was maintained in cellobiose culture. The gene response of bgl1B is not only similar to that of the intracellular BGL gene (bgl2) in Trichoderma reesei (Saloheimo et al. 2002), but also resembles those of other cellulolytic enzymes whose expressions are regulated by carbon catabolite repression. In a previous study of intracellular BGLs from P. chrysosporium, the production patterns of two BGLs were compared in glucose and cellobiose cultures (Smith and Gold 1979). In that work, one intracellular BGL production was induced by cellobiose and repressed by glucose, whereas the response of another intracellular BGL production to carbon source was slow. These production patterns are similar to the gene responses of BGL1B and BGL1A, respectively, in the present study. Considering that the transcription of bgl3A is repressed in both glucose and cellobiose cultures, as reported previously, only bgl1B positively responds to cellobiose among the three BGLs.
A comparison of the kinetics and gene responses of the three BGLs from P. chrysosporium (BGL1A, BGL1B, and BGL3A) indicates that BGL1B may be responsible for cellobiose. For this to occur, however, cellobiose should be transported from extracellular to intracellular regions, contrary to the formerly proposed pathway, in which cellobiose is hydrolyzed to glucose by extracellular enzymes. In addition, because the affinity of CDH for cellobiose (K m=0.0432 mM) is still comparable with that of BGL1B (Yoshida et al. 2001), and BGL1B also hydrolyzes cellobionolactone (K m=1.07±0.07 mM, k cat=3.05±0.06 s−1), both cellobiose and cellobionolactone may serve as substrates for BGL1B. Further investigations, such as transgenic and metabolomic analysis, will be necessary to establish how the fungus utilizes cellulosic substrates.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ayers AR, Ayers SB, Eriksson KE (1978) Cellobiose oxidase, purification and partial characterization of a hemoprotein from Sporotrichum pulverulentum. Eur J Biochem 90:171–181
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: signalP 3.0. J Mol Biol 340:783–795
Broda P, Birch PR, Brooks PR, Sims PF (1995) PCR-mediated analysis of lignocellulolytic gene transcription by Phanerochaete chrysosporium: substrate-dependent differential expression within gene families. Appl Environ Microbiol 61:2358–2364
Eriksson KE (1978) Enzyme mechanisms involved in cellulose hydrolysis by rot fungus Sporotrichum pulverulentum. Biotechnol Bioeng 20:317–332
Eriksson KE, Pettersson B, Westermark U (1974) Oxidation: an important enzyme reaction in fungal degradation of cellulose. FEBS Lett 49:282–285
Habu N, Igarashi K, Samejima M, Pettersson B, Eriksson KE (1997) Enhanced production of cellobiose dehydrogenase in cultures of Phanerochaete chrysosporium supplemented with bovine calf serum. Biotechnol Appl Biochem 26:97–102
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Henrissat B, Bairoch A (1993) New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 293:781–788
Henrissat B, Bairoch A (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem J 316:695–696
Igarashi K, Samejima M, Saburi Y, Habu N, Eriksson KE (1997) Localization of cellobiose dehydrogenase in cellulose-grown cultures of Phanerochaete chrysosporium. Fungal Genet Biol 21:214–222
Igarashi K, Tani T, Kawai R, Samejima M (2003) Family 3 β-glucosidase from cellulose-degrading culture of the white-rot fungus Phanerochaete chrysosporium is a glucan 1,3-β-glucosidase. J Biosci Bioeng 95:572–576
Johnsrud SC, Eriksson KE (1985) Cross-breeding of selected and mutated homokaryotic strains of Phanerochaete chrysosporium K-3—new cellulase deficient strains with increased ability to degrade lignin. Appl Microbiol Biotechnol 21:320–327
Kawai R, Yoshida M, Tani T, Igarashi K, Ohira T, Nagasawa H, Samejima M (2003) Production and characterization of recombinant Phanerochaete chrysosporium β-glucosidase in the methylotrophic yeast Pichia pastoris. Biosci Biotechnol Biochem 67:1–7
Kawai R, Igarashi K, Kitaoka M, Ishii T, Samejima M (2004) Kinetics of substrate transglycosylation by glycoside hydrolase family 3 glucan (1->3)-β-glucosidase from the white-rot fungus Phanerochaete chrysosporium. Carbohydr Res 339:2851–2857
Kawai R, Igarashi K, Yoshida M, Kitaoka M, Samejima M (2005) Hydrolysis of β-1,3/1,6-glucan by glycoside hydrolase family 16 endo-1,3(4)-β-glucanase from the basidiomycete Phanerochaete chrysosporium. Appl Microbiol Biotechnol (in press) https://doi.org/10.1007/s00253-005-0214-4
Kremer SM, Wood PM (1992) Evidence that cellobiose oxidase from Phanerochaete chrysosporium is primarily an Fe(III) reductase—kinetic comparison with neutrophil NADPH oxidase and yeast flavocytochrome b 2. Eur J Biochem 205:133–138
Li B, Renganathan V (1998) Gene cloning and characterization of a novel cellulose-binding β-glucosidase from Phanerochaete chrysosporium. Appl Environ Microbiol 64:2748–2754
Lymar ES, Li B, Renganathan V (1995) Purification and characterization of a cellulose-binding β-glucosidase from cellulose-degrading cultures of Phanerochaete chrysosporium. Appl Environ Microbiol 61:2976–2980
Martinez D, Larrondo LF, Putnam N, Gelpke MD, Huang K, Chapman J, Helfenbein KG, Ramaiya P, Detter JC, Larimer F, Coutinho PM, Henrissat B, Berka R, Cullen D, Rokhsar D (2004) Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat Biotechnol 22:695–700
Mchale A, Coughlan MP (1981a) The cellulolytic system of Talaromyces emersonii—identification of the various components produced during growth on cellulosic media. Biochim Biophys Acta 662:145–151
Mchale A, Coughlan MP (1981b) The cellulolytic system of Talaromyces emersonii—purification and characterization of the extracellular and intracellular β-glucosidases. Biochim Biophys Acta 662:152–159
Nielsen H, Engelbrecht J, Brunak S, von Heijne G (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng 10:1–6
Peralta RM, Terenzi HF, Jorge JA (1990) β-D-Glycosidase activities of Humicola grisea: biochemical and kinetic characterization of a multifunctional enzyme. Biochim Biophys Acta 1033:243–249
Saloheimo M, Kuja-Panula J, Ylosmaki E, Ward M, Penttila M (2002) Enzymatic properties and intracellular localization of the novel Trichoderma reesei β-glucosidase BGLII (cel1A). Appl Environ Microbiol 68:4546–4553
Smith MH, Gold MH (1979) Phanerochaete chrysosporium β-glucosidases: induction, cellular localization, and physical characterization. Appl Environ Microbiol 37:938–942
Takashima S, Nakamura A, Masaki H, Uozumi T (1996) Purification and characterization of cellulases from Humicola grisea. Biosci Biotechnol Biochem 60:77–82
Takashima S, Nakamura A, Hidaka M, Masaki H, Uozumi T (1999) Molecular cloning and expression of the novel fungal β-glucosidase genes from Humicola grisea and Trichoderma reesei. J Biochem (Tokyo) 125:728–736
Tempelaars CA, Birch PR, Sims PF, Broda P (1994) Isolation, characterization, and analysis of the expression of the cbhII gene of Phanerochaete chrysosporium. Appl Environ Microbiol 60:4387–4393
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Westermark U, Eriksson KE (1974) Cellobiose: quinone oxidoreductase, a new wood-degrading enzyme from white-rot fungi. Acta Chem Scand B 28:209–214
Yoshida M, Ohira T, Igarashi K, Nagasawa H, Aida K, Hallberg BM, Divne C, Nishino T, Samejima M (2001) Production and characterization of recombinant Phanerochaete chrysosporium cellobiose dehydrogenase in the methylotrophic yeast Pichia pastoris. Biosci Biotechnol Biochem 65:2050–2057
Yoshida M, Igarashi K, Kawai R, Aida K, Samejima M (2004) Differential transcription of β-glucosidase and cellobiose dehydrogenase genes in cellulose degradation by the basidiomycete Phanerochaete chrysosporium. FEMS Microbiol Lett 235:177–182
Acknowledgements
The authors are grateful to Dr. Shinya Fushinobu (Department of Biotechnology, University of Tokyo) for valuable discussions about the kinetics of GH family 1 BGL. This research was supported by a Grant-in-Aid for Scientific Research to M. Samejima (number 17380102) from the Japanese Ministry of Education, Culture, Sports and Technology.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Tsukada, T., Igarashi, K., Yoshida, M. et al. Molecular cloning and characterization of two intracellular β-glucosidases belonging to glycoside hydrolase family 1 from the basidiomycete Phanerochaete chrysosporium . Appl Microbiol Biotechnol 73, 807–814 (2006). https://doi.org/10.1007/s00253-006-0526-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0526-z