
Abstract
A heat-stable sarcosine oxidase produced by Bacillus sp. BSD-8 (SOX) had been studied and its complete gene sequence, which contained 1,164 bp nucleotides and encoded a protein of 387 amino acids, was obtained by DNA Walking method. The sox gene was cloned and functionally overexpressed in E. coli and the recombinant SOX (rSOX) was purified to homogeneity, its properties was studied and compared with the wild type of SOX. The rSOX as well as SOX was stable at 60°C and at pH 7.0∼10.0, respectively. The optimal temperature for this enzyme was 60°C and at pH 8.5, it showed its highest activity. The Km and Kcat of the enzyme was 3.1 mM and 20.3/s, respectively. The difference between the properties of the SOX and rSOX was that the SOX contained noncovalent FAD, whereas the rSOX contained covalent FAD. The study also showed that an increased number of alanine residues in the rSOX might have some contribution in the enzymatic thermostability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sarcosine oxidase (SOX, EC 1.5.3.1) catalyzes the oxidative demethylation of sarcosine and forms equimolar of formaldehyde, glycine and hydrogen peroxide (Suzuki 1981). Many organisms have been found to produce intracellular SOX, such as bacteria (Kim et al. 1987; Matsuda et al. 1987; Mori et al. 1980; Nishiya and Imanaka 1993; Ogushi et al. 1988; Suzuki 1981), mammals (Chikayama et al. 2000; Reuber et al. 1997), and even plant (Goyert et al. 2004). This enzyme, together with the other enzymes such as creatininase and creatinase, have been used in clinics for determination of creatinine, which is an important parameter to estimate the function of kidney (Bacon and Pardue 1991; Kinoshita and Hiraga 1980; Lindback and Bergman 1989; Suzuki 1994). In general, these enzymatic methods are based on the following reactions:

The quantity of hydrogen peroxide produced in the final reaction was directly proportional to the creatinine of the sample. By measuring the H2O2, the concentration of creatinine can be determined.
At present, several SOXs from microorganisms have been studied (Chlumsky et al. 1993; Matsuda et al. 1987; Mori et al. 1980; Ogushi et al. 1988; Suzuki 1981), and even a few SOXs, such as those from Streptomyces sp. KB210-8SY (Suzuki et al. 1992), Bacillus sp. NS-129 (Kayama et al. 1991), and Bacillus sp. B-0618 (Suzuki et al. 1994), have been cloned and expressed. It was found that three types of SOXs could be distinguished on the basis of their quanternary structure: monomers, heterodimers, and heterotetramers. Among them, monomeric SOX contains covalently bound flavin (Willie et al. 1996), heterotetrameric SOX contains covalently or noncovalently bound FAD and NAD+ and covalently bound FMN (Trichey et al. 1999; Willie et al. 1996; Willie and Jorns 1995). At present, monomeric enzymes are mainly applied for clinical assays.
As an enzyme for clinical applications, thermostability is an important property. An enzyme with the property of high thermal stability will have advantages in transportation, storage, and clinical application. In previous work, a Bacillus sp. BSD-8 that produced thermostable SOX was isolated in our laboratory and this enzyme had been purified and systematically studied. It was shown that the enzyme had a relatively high thermostability and may have advantage in the clinical application for measurement of creatinine.
In this study, the cloning and amino acid sequence study of the Bacillus sp. BSD-8 SOX gene are reported. The procedures for the overexpression of the recombinant SOX (rSOX) in E. coli, its purification, and characterization are also described.
Materials and methods
Bacterial strains, plasmids, and culture conditions
Bacillus sp. BSD-8, as a source of SOX gene, was isolated by our lab, E. coli JM109 and E. coli BL21 (DE3) were used as the hosts for plasmid amplification and expression, respectively. Plasmid pBluescript II KS (−) and pET-15b (−) were used as vectors.
Bacillus sp. BSD-8 was incubated at 30°C for 24 h in a shaker at 120 rpm in a medium which contained 1% (w/v) creatine, 0.8% (w/v) yeast extract, 0.3% (w/v) NaCl, 0.05% (w/v) MgSO4·7H2O, 0.2% (w/v) K2HPO4, and 0.05% (w/v) KH2PO4·3H2O, pH 7.0. The E. coli and recombinant strains were grown in Luria–Bertani (LB) medium at 37°C overnight with vigorous shaking. When necessary, ampicillin (100 μg/ml) was added to the media.
Reagents and supplies
Restriction enzymes, Taq DNA polymerase, T4 DNA ligase, and Gel extraction kit were obtained from Takara, Japan. Oligonucleotides were synthesized from BioAsia (China). PGEM-T vector, expression vector pET15b, Ni-iminodiacetic acid (IDA) resin, and thrombin were purchased from invitrogen. All other reagents were purchased from Hangzhou Huadong Medicine Goup (Hangzhou, China).
Polymerase chain reaction-mediated chromosome walking
Genomic DNA of Bacillus sp. BSD-8 was isolated following the method described by Sambrook and Russell (2001). The sequences of the oligonucleotide primers used in this study are summarized in Table 1. With the genomic DNA of Bacillus sp. BSD-8 as the template, a first polymerase chain reaction (PCR) was performed with gene-specific primers, Up1 and Down1, which were designed according to the strongly conserved amino acid sequence of SOX from Bacillus sp. B-0618 (GenBank no. D16521) and Bacillus sp. NS-129 (GenBank no. D10553) using Primer Premier 5.0 software. The PCR products were separated by electrophoresis in 1.5% agar gels, cloned into the PGEM-T vector (Invitrogen) according to standard methods (Sambrook and Russell 2001).
To obtain the 3′ portion of the SOX sequence, the genome walking method was applied, Bacillus sp. BSD-8 genomic DNA was completely digested with PstI, followed by purification and ligation with the daptor at the 1:10 mol ratio, which was synthesized with two oligonucleotides, AP1 and AP2, with the method described by Slebert et al. (1995). A second PCR was carried out using primers SDP2 and DAP, which were designed with first PCR product and the adaptor, respectively. Both of the PCRs were performed at 94°C for 1 min, 52°C for 1 min, and 72°C for 1 min for 32 cycles using Eppendorf Master Cycler Gradient (Brinkmann Instruments, NY, USA).
DNA sequence analysis
DNA sequencing was carried out with the dideoxy-chain termination method (Sanger et al. 1997) by using an ABI Prism 377 Genetic Analyser (Applied Biosystems, California, USA). DNAStar (Lasergene) and OMIGA2.0 softwares were employed to analyze the DNA sequence.
Cloning of the SOX gene from Bacillus sp. BSD-8 and overexpression of rSOX in E. coli
Standard DNA manipulation techniques were used for isolation of plasmid DNA, restriction digestion, ligation, and transformation (Sambrook and Russell 2001).
The SOX gene was amplified by PCR from BSD-8 using primers P1 and P2 (Table 1). The primers had noncomplementary 5′ end and 3′ end carrying XhoI and BamHI restriction sites (underlined), respectively. The purified PCR product was digested with XhoI and BamHI and ligated with pET15b (−) digested with the same restriction enzymes. The expression vector for rSOX was used to transform E. coli strain BL21(DE3). To express the rSOX, the transformant was cultivated in a 100-ml LB medium containing 100 μg/ml ampicillin at 37°C to an OD600 of 0.6 and induced at 30°C by 1 mM of IPTG for another 5 h on shaking incubator at 250 rev min−1.
Purification of His6-tagged rSOX and removal of the tag
After cultivation, the cells were collected by centrifugation at 2,750×g at 4°C for 15 min, washed twice with binding buffer (20 mM Tris–HCL, 500 mM NaCl, and 5 mM imidazole, pH 7.9) and lysed by sonication. The cell debris was then removed by centrifugation at 2,750×g at 4°C (Ichikawa et al. 1993) for 30 min, and the supernatant was loaded onto a 2.5-ml Ni-IDA column (invitrogen) preequilibrated with binding buffer. The column was subsequently washed with 15 ml binding buffer and washing buffer (20 mM Tris–HCL, 500 mM NaCl, and 10 mM imidazole, pH 8.0) until no more protein was eluted. The rSOX was then eluted with 20 ml eluting buffer (20 mM Tris–HCL, 500 mM NaCl, and 100 mM imidazole).
To remove His-tag from recombinant enzyme, the enzyme solution, which contained 6 mg of purified rSOX was added with 2.5 mM CaCl2 and 50 U thrombin, reacted at 4°C for 3 h. The uncut protein and the His6 fragment were removed by Ni-IDA resin filtration and the rSOX was concentrated by precipitation with 70% saturated (NH4)2SO4 and dialyzed against 50 mM phosphate buffer at pH 7.5.
The molecular mass of rSOX and the purity of the enzyme in the process of purification were analyzed by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Coomassie brilliant blue. Protein concentration was determined by the method of Bradford 1976.
SOX assay
To measure the SOX activity, 0.1 ml enzyme solution was added in to a 0.9-ml sodium pyrophosphate buffer (pH 8.0) containing 0.1 M sarcosine as substrate. After 10 min incubation at 37°C, the reaction >was terminated with 0.25 ml of 1 M acetic acid. Formaldehyde formed in this reaction was measured by the method of Nash (Peter et al. 1997). Briefly, the reaction mixture (1.25 ml) was incubated with 1.5 ml of 20% ammonium acetate containing 0.02% acetylacetone at 37°C for 40 min and the absorbance at 410 nm was read against the blank. One unit of enzyme activity was defined as the amount of the enzyme that catalyzed the oxidation of 1 μmol of substrate per minute under the conditions described above.
Characterization of the rSOX
To determine the effect of temperature on the rSOX activity, the enzyme activity was measured at various temperatures (20–65°C) for 10 min. For determination of the thermostability of the enzyme, the enzyme solution was incubated at 20–65°C for 10 and 30 min, respectively, and the remaining activity was determined by standard method. The optimum pH range for rSOX reaction was estimated by using the following buffers: 0.2 M citrate for pH 4–6; 0.2 M Tris–HCl for pH 6–8; 0.2 M boric acid–Na borate for pH 8–9.5, and 0.2 M boric acid–NaOH for pH 9.5–11. For the pH stability assay, the enzyme was incubated in the above buffers at 25°C for 18 h; the remaining activity was then measured by standard method. For kinetic study, substrate concentrations were varied between 1 and 8 mM and the Michaelis constant was evaluated by Lineweaver–Burk plots method.
Prosthetic groups of SOX and rSOX
Following the method described by S. Ogushi et al. (1988), both of the purified SOX and rSOX were dissolved in sodium pyrophosphate buffer, pH 8.0, and kept in a boiling water bath for 10 min in the presence of 10% trichloroacetic acid to denature the enzyme, centrifuged at 14,000×g for 5 min. The noncovalently bound flavin then was released to solution upon denaturation.
Nucleotide accession number
The nucleotide sequence of the SOX gene was deposited under the accession number AY626822.
Results
Isolation of the sox gene from Bacillus sp. BSD-8
With genomic DNA as the template, PCR was done by using primer Up1/Down1, and two major bands, about 1,000 and 270 bp in length, in 1.5% agar gel were recovered and sequenced. A complete BLAST homologous search in the GenBank database showed that the larger fragment had a high similarity with the 5′ upstream region of the some other SOX genes, therefore it was used for designing new gene-specific primer SDP2 for genome walking experiment. Genome walking library was constructed by ligation of the adaptor with PstI-digested genomic DNA and it was used as template to isolate the 3′ downstream regions of the SOX gene. Downstream genome walking was performed with DAP and SDP2 as primers and a notable 750 bp band was amplified. It was sequenced, and a BLAST retrieve found its SOX gene homologous sequence and overlap in its N terminus with the previously obtained fragment. The SOX gene was then assembled according to overlapping sequences from the two fragments by using DNAStar software.
The nucleotide sequence of the gene and the deduced amino acid sequence of its protein product were deposited in GenBank (accession no. AY626822). The gene has an open reading frame of 1,164 bp and codes for a polypeptide of 387 residues with a molecular mass of 43 kDa predicted by the gene sequence.
Sequence comparison of Bacillus sp. BSD-8 SOX with its bacterial counterparts
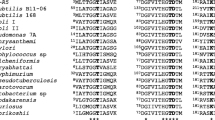
The sequence of BSD-8 SOX gene showed significant homology with the other monomeric SOX sequences present in NCBI. The deduced amino acid sequence of Bacillus sp. BSD-8 SOX showed maximum identity with SOX from Bacillus sp. NS-129 (91%); in addition, it showed 80, 82, and 82% identity with SOXs from Bacillus sp. B-0618, Arthrobacter. sp. TE1826, and Bacillus sp. KS-11A, respectively. Sequence alignment of the deduced amino acid sequence of the SOX of Bacillus sp. BSD-8 with those from Bacillus sp. B-0618, Arthrobacter. sp. TE1826 (GenBank no. D63413), Bacillus sp. NS-129, and Bacillus sp. KS-11A (GenBank no. NP_014683) revealed the presence of highly conserved residues (Fig. 1).

Sequence alignment of SOXs of Bacillus sp. BSD-8, Bacillus sp.NS-129, Bacillus sp. B-0618, Bacillus sp. KS-11A, and Arthrobacter. sp.TE1826. Sequences were compared using OMIGA2.0 software. Identical residues and conserved substitutions are shadowed black and gray, respectively. The 14 amino acid residues meet that criterion are indicated by rectangles
Expression and purification of rSOX
The rSOX was successfully expressed in transformed E. coli strain BL21(DE3) with the pET-15b vector. After induction with IPTG, the E. coli cells were collected from LB medium, and a cell lysate was prepared. The recombinant protein was further purified by using metal-chelate affinity chromatography (Ni2+-IDA column). With the 6× His-tag at the N terminus, which facilitated strong binding of the protein to the Ni-IDA matrix, most of the cytosolic proteins were washed away from the column with the buffer containing 10 mM imidazole. The purity of the enzyme was examined by SDS-PAGE and a clear single band corresponding to the 51 kDa protein was observed (Fig. 2). The recombinant protein was overexpressed to more than 30% total cell protein. About 38.4 mg highly purified rSOX from E. coli cells with an approximate 32-fold purification was obtained. The specific activity of the purified enzyme was 28.6 U/mg protein when sarcosine was used as the substrate.

SDS-PAGE analysis of total-cell lysate and purified protein. Lane 1 uninduced protein sample. Lane 2 induced protein sample. Lane 3 Ni2+–IDA purified rSOX protein sample (corresponds to 51 kDa). Lane M protein molecular weight marker
The properties of rSOX
The expressed rSOX had optimal activity at a temperature of 60°C (Fig. 3a). The activity rose in an almost linear fashion from 25 to 60°C, but decreased abruptly once the temperature exceeded 65°C, with only 13.5% activity remaining at 65°C. It was shown that the rSOX was stable when incubation at temperatures below 60°C (Fig. 3b). The rSOX expressed in E. coli had an optimal pH of 8.5 in the assay buffer (Fig. 4a). The pH-stability of rSOX was examined by incubation at 25°C at various pH for 18 h and measurement of residual activity. The rSOX was stable at the pH range from 6.5 to 11.0 (Fig. 4b). At optimal pH and temperature, the Km and the Kcat were estimated to be 3.1 mM and 20.3/s, respectively.

Effect of temperature on enzyme activity and stability. a The effect of temperature on rSOX activity was determined at various temperatures, as described in the text. b The effect of temperature on rSOX stability was determined by incubating the pure rSOX at various temperatures for 10 and 30 min, respectively, and measuring the remaining activity. (▪) 10 min; (□) 30 min

Effect of pH on enzyme activity and stability. a Activity was measured at 37°C at various pHs. b rSOX was preincubated at various pHs and 25°C for 18 h, and its residual activity was measured at 37°C. (▲) pH 4–6, 0.2 M citrate buffer; (□) pH 6–8, 0.2 M Tris–HCl buffer; (▪) pH 8–9.5, 0.2 M boric acid–Na borate buffer; (□) pH 9.5–11, 0.2 M boric acid–NaOH buffer
Prosthetic groups of SOX and rSOX
Both of the purified SOX and rSOX had a yellow color, and their sarcosine oxidizing activity was not dependent on FAD addition. When the rSOX was kept in a boiling water bath for 10 min in the presence of 10% trichloroacetic acid, the supernatant of rSOX obtained by centrifugation did not contain any colored material, whereas the protein pellet was yellow, suggesting the existence of covalently bound flavin and the flavin was tightly associated with the protein. However, same treatment of the SOX gave a yellow-colored material in the soluble fraction, suggesting the existence of noncovalently bound flavin in that wild type SOX.
Discussion
The wild type SOX from Bacillus sp. BSD-8 had been purifed by ammonium sulfate precipitation, DEAE-cellulose ion exchange, hydrophobic and molecular sieve chromatography, and the purified rSOX shared same properties with that wild type SOX protein, including optimum pH, pH stability, maximal temperature, thermostability, and Km values (data not shown). However, they were different in flavin binding mode: rSOX presumably contained covalently bound flavin and SOX contained noncovalently bound flavin. This might be due to the different host systems (SOX from Bacillus sp. BSD-8 and rSOX from E. coli) resulting in the different flavin binding mode to protein.
Thermostability has become a desirable characteristic for many enzymes. At present, the structural features responsible for the thermostability of SOX, such as amino acid residues involved in the thermostabilization, are still unknown. Because the SOXs from Bacillus sp. BSD-8 and other strains had different thermostability in the range from 30 to 60°C and their amino acids sequences have been available at present, it provides a basis for the analysis of the relationship between the structure and thermostability of this enzyme.
Based on the alignment analysis among five SOXs in Fig. 2, the SOXs from strain BSD-8 and NS-129 are the two most similar enzymes, which also were two most thermostable enzymes. As indicated in Table 2 (Ichikawa et al. 1993; Suzuki 1988; Matsuda et al. 1987; Nishiya and Imanaka 1993) and Fig. 1, the SOX from strain BSD-8 was still 5°C more thermostable than that from NS-129, and there are 32 amino acid residues different between them. Considering that only very little part of mutations of amino acid residues would change the thermostability of an enzyme and the SOX from strain BSD-8 was the most thermostable one, among these 32 amino acid residues, the amino acid residues that promote the thermostability of SOX of strain BSD-8 most probably would also differ from the amino acid residues of the other SOXs at equivalent position. As shown in Fig. 1, there are only 14 amino acid residues meeting this criterion and among them, alanine took a considerable part: there are six alanines mutated from other amino acid residues. This phenomenon imply that there would be some relationship between composition of alanine of SOX and its thermostability. It was also found that in the Glucose-1-phosphate thymidylyltransferase from Thermuscaldophilus, a thermophilic enzyme, contained relative higher level of alanine than its mesophilic counterparts (Parajuli et al. 2005). Borgi et al. (2004) found that in the glucose isomerase of the Streptomyces sp., the presence of an Ala residue at position 103 was especially important for its thermostability and the Ala103Gly mutation introduced in glucose isomerase could significantly decrease the half-life time of that enzyme at 90°C.
Generally, the thermal stability of protein is increased with an increase in hydrophobicity (Yutani et al. 1987), helix stability (Dao-Pin et al. 1990; Imanaka et al. 1986), and tight packing and ionic interactions of the enzymes (Lim et al. 2001; Vetriani et al. 1998). It was thought that the large number of alanine residues, which are found frequently in helices (Branden and Tooze 1991) and was thought to enhance the helix packing, thereby increased protein rigidity (Argos et al. 2006; Menendez-Arias and Argos 2006) and their thermostability. In addition, an increase in the number of glutamic acid residues of SOX of strain BSD-8, which was able to interact with more distant residues in the enzyme while simultaneously increasing protein surface hydrophobicity (Argos et al. 2006), might also had some contribution in the thermostablility of SOX.
References
Argos P, Rossmann MG, Grau UM, Zuber H, Frank G, Tratschin JD (2006) Thermal stability and protein structure. Biochemistry 18:5698–5703
Bacon BL, Pardue HL (1991) Predicitive error-compensating kinetic method for enzymetic quantification of creatinine in serum. Clin Chem 37:1338–1344
Borgi MA, Srih-Belguith K, Ben M, Mezghani M, Tranier S, Haser R, Bejar S (2004) Glucose isomerase of the Streptomyces sp. SK strain: purification, sequence analysis and implication of alanine 103 residue in the enzyme thermostability and acidotolerance. Biochimie 86:561–568
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein. Anal Biochem 72:248–254
Branden C, Tooze J (1991) Introduction to protein structure. Garland, New York
Chikayama M, Ohsumi M, Yokota S (2000) Enzyme cytochemical localization of sarcosine oxidase activity in the liver and kidney of several mammals. Histochem Cell Biol 113:489–495
Chlumsky LJ, Zhang L, Ramsey AJ, Jorns MS (1993) Preparation and properties of recombinant corynebacterial sarcosine oxidase: evidence for posttranslational modification during turnover with sarcosine. Biochemistry 32:11132–11142
Dao-Pin S, Baase WA, Matthews BW (1990) A mutant T4 lysozyme (Val 131 Ala) designed to increase thermostability by the reduction of strain within an alpha-helix. Proteins 7:198–204
Goyert A, Johnson TL, Olsen LJ, Collakova E, Hill YS, Rhodes D, Hanson AD (2004) Characterization and metabolic function of a peroxisomal sarcosine and pipecolate oxidase from Arabidopsis. J Biol Chem 279:16947–16953
Ichikawa T, Suzuki M, Koyama T (1993) Japan Patent 1993115281-A1
Imanaka T, Shibazaki M, Takagi M (1986) A new way of enhancing the thermostability of proteases. Nature 324:695–697
Kayama Y, Yamamoto H, Suzuki M, Nakano E (1991) Cloning and espression of the sarcosine oxidase gene from Bacillus sp.NS-129 in Escherichia coli. Agric Biol Chem 55:1259–1263
Kim JM, Shimizu S, Yamada H (1987) Crystallization and characterization of sarcosine oxidase from Alcaligenes denitrificans subp.denitrificans. Agric Biol Chem 51:1167–1168
Kinoshita T, Hiraga Y (1980) A fluorophotometric determination of serum creati-nine and creatine using a creatine amidohydrelase–creatine amidinohydrolase–sarcosine oxidase–peroxidase system and diacetyldichloroflurorescin. Chem Pharm Bull 28:3501–3506
Lim JH, Hwang KY, Choi J, Lee DY, Ahn BY, Cho Y, Kim KS, Han YS (2001) Mutational effects on thermostable superoxide dismutase from Aquifex pyrophilus: understanding the molecular basis of protein thermostability. Biochem Biophys Res Commun 288: 263–268
Lindback B, Bergman A (1989) A new commercial method for the enzymetic determination of creatine in serum and urine evaluated: comparision with the kinetic Jaffe method and isotope dilution-mass spectrometry. Clin Chem 25:835–837
Matsuda Y, Hoshika H, Inouye Y, Ikut S, Matsuura K, Nakamura S (1987) Purification and characterization of sarcosine oxidase of Bacillus origin. Chem Pharm Bull 35:711–717
Menendez-Arias L, Argos P (2006) Engineering protein thermal stability: sequence statistics point to residue substitutions in a-helices. J Mol Biol 206:397–406
Mori N, Sano M, Tani Y, Yamda H (1980) Purification and properties of sarcosine oxidase from Cylindrocarpon didymium M-1. Agric Biol Chem 44:1391–1397
Nishiya Y, Imanaka T (1993) Cloning and sequencing of the sarcosine oxidase gene from Arthrobacter sp.TE1826. J Ferment Bioeng 75:239–244
Ogushi S, Nagao K, Emi S, Ando M, Tsuru D (1988) Sarcosine oxidase from Arthrobacter ureafaciens: purification and some properties. Chem Pharm Bull 36:1445–1450
Parajuli N, Basnet DB, Chung YS, Lee HC, Liou K, Liou K (2005) Biochemical characterization of a novel thermostable glucose-1-phosphate thymidylyltransferase from Thermuscaldophilus: probing the molecular basis for its unusual thermostability. Enzyme MicrobTechnol 37:402–409
Peter B, Josef A, Klaus DK, Ralf M (1997) Cloning, nucleotide sequence and expression of a mannitol dehydrogenase gene from Pseudomonas fluorescens DSM 50106 in Escherichia coli. Biochim Biophys Acta 1351:157–167
Reuber BE, Karl C, Reimanm SA, Mihalik SJ, Dodt G (1997) Cloning and functional expression of a mammalian gene for a peroxisomal sarcosine oxidase. J Biol Chem 272:6766–6776
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
Sanger F, Nicklen S, Coulson AR (1997) DNA sequencing with chain terminating inhibitors. Proc Natl Acad Sci 74:5463–5467
Slebert PD, Chenchik A, Kellogg DE (1995) An improved PCR method for walking in uncloned genomic DNA. Nucleic Acids Res 23:1087–1088
Suzuki M (1981) Purification and some properties of sarcosine oxidase from Corynebacterium sp. U-96. J Biochem 89:599–607
Suzuki M (1988) US Patent 4,740,465
Suzuki H (1994) Sarcosine oxidase: structure, function, and the application to creatinine determination. Amino Acids 7:27–43
Suzuki K, Ogishima M, Sugiyama M, Inouye Y, Nakamura S, Imamura S (1992) Molecular cloning and expression of a Streptomyces sarcosine oxidase gene in Streptomyces lividans. Biosci Biotechnol Biochem 56:432–436
Suzuki K, Sagai H, Sugiyama M (1994) Cloning, sequencing and overexpression in Escherichia coli of a sarcosine oxidase-encoding gene linked to the Bacillus creatinase gene. J Ferment Bioeng 77:231–234
Trichey P, Wagner MA, Jorns MS, Mathews FS (1999) Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure Fold Des 7:331–345
Vetriani C, Maeder DL, Tolliday N, Yip KS, Stillman TJ, Britton KL, Rice DW, Klump HH, Robb FT (1998) Protein thermostability above 100°C: a key role for ionic interactions. Proc Natl Acad Sci USA 95:12300–12305
Willie A, Jorns MS (1995) Discovery of a third coenzyme in sarcosine oxidase. Biochemistry 34:16703–16707
Willie A, Edmondson DE, Jorns MS (1996) Sarcosine oxidase contains a novel covalently bound FMN. Biochemistry 35:5292–5299
Yutani K, Ogasahara K, Tsujita T, Sugino Y (1987) Dependence of conformational stability on hydrophobicity of the amino acid residue in a series of variant proteins substituted at a unique position of tryptophan synthase subunit. Proc Natl Acad Sci USA 81:4441–4444
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guo, K., Ma, X., Sun, G. et al. Expression and characterization of a thermostable sarcosine oxidase (SOX) from Bacillus sp. in Escherichia coli . Appl Microbiol Biotechnol 73, 559–566 (2006). https://doi.org/10.1007/s00253-006-0502-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-006-0502-7