
Abstract
A serine protease produced by Thermomonospora fusca YX (TfpA) is heat-stable (up to 85°C) and has a broad pH activity range and strong resistance to detergents. The objective of this study was to determine if the methylotropic yeast Pichia pastoris could express TfpA extracellularly. A 1.0-kb DNA fragment (tfpA) encoding the pro-peptide and mature protein of TfpA was cloned into expression vectors pPICZαA (inducible) and pGAPZαA (constitutive) and introduced into P. pastoris by electroporation. Expression of r-TfpA was greater in the inducible system than in the constitutive one, producing 135 U ml−1 medium supernatant 6 days after methanol induction. The r-TfpA was not glycosylated (21.7 kDa), and had pH and temperature optima of 8.5 and 80°C, respectively, using azocasein as a substrate. In conclusion, P. pastoris can be used as a host to produce extracellular r-TfpA, and expression efficiency may be improved by optimizing fermentation conditions and modifying factors related to protein expression and stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteases constitute one of the most important groups of industrial enzymes (Layman 1986). In particular, alkaline proteases have many applications in the detergent, protein, brewing, meat processing, photographic, leather and dairy industries (Anwar and Saleemuddin 1998). For most of these applications, it is ideal that a protease has good stability for processing and storage, high catalytic efficiency over a broad pH range and substrate specificity, and resistance to detergents such as sodium lauryl sulfate (SDS). Many researchers have studied alkaline proteases from various sources such as bacteria or fungi (Anwar and Saleemuddin 1998; Kumar and Takagi 1999); however, most of these enzymes do not satisfy all the above requirements.
A serine protease from Thermomonospora fusca is of great potential. As a filamentous soil bacterium, T. fusca actively degrades cellulose and other plant cell wall polymers in moderately thermophilic environments (Wilson 1988). Likewise, the serine protease produced by this organism is fairly thermostable. The enzyme belongs to the chymotrypsin family, and cleaves near hydrophobic amino acids (Gusek and Kinsella 1987; Kristjansson and Kinsella 1990). It is first synthesized as a 375-residue pre-pro-protein and is cleaved to produce the 194-residue active mature protease (Lao and Wilson 1996). This enzyme is also resistant to detergents and shows a broad pH range and substrate specificity, with a preference for activity against aromatic and hydrophobic amino acids.
Lao and Wilson (1996) expressed the protease gene (tfpA) in Streptomyces lividans as an active extracellular protease. However, the recombinant protease was precipitated and inactivated at ambient temperature due to an inherent protease inhibitor. Although a protease-inhibitor mutant strain might be used to avoid the precipitation problem, S. lividans is still not an ideal host due to the poor yield. In contrast, the methylotropic yeast Pichia pastoris has been used successfully for expression of heterologous proteins including phytase and keratinolytic protease (Higgins and Cregg 1988; Rodriguez et al. 2000; Porres et al. 2002). Therefore, our objective was to determine if an inducible or constitutive expression system of P. pastoris could be used to express tfpA as an active, extracellular enzyme.
Materials and methods
DNA amplification and cloning
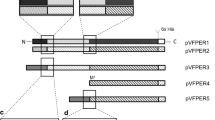
The full sequence of the tfpA gene from T. fusca YX (TfpA, signal peptide, pro-peptide, and mature protein) was cloned into the plasmid pUC19 (Lao and Wilson 1996). The sequence containing the pro-peptide and mature protein was amplified by PCR using pUC19 as the template. Primer tfp1 (forward: 5′-CGGAATTCCAAGAGCTCGCCCT CAAACGC-3′) was designed to amplify pro- and mature protein from the gene and to generate an EcoRI site at the 5′ end. Primer tfp2 (reverse: 5′-GCTCTAGATCAGCCGGTGAC CAGTTG-3′) was designed to generate an XbaI site at the 3′ end. The thermal program used to amplify the sequence included 1 cycle at 94°C (3 min), 30 cycles of [94°C (1 min), 59°C (1 min) and 72°C (1.5 min)], and 1 cycle at 72°C (10 min). PCR reactions were carried out in a Gene Amp PCR system 2400 (Perkin Elmer, Norwalk, Conn.) using the Thermo-Taq system kit (Takara, Kyoto, Japan). Amplified PCR products were resolved by 1% low melting temperature agarose gel electrophoresis. A gel slice containing the expected band (1 kb) was excised and the DNA was eluted using a gel extraction kit (BIO101, Carlsbad, Calif.). The PCR product was cloned into the vector pGEM-T (Promega, Madison, Wis.) and introduced into Escherichia coli JM109 by electroporation, followed by screening for positive colonies. The isolated fragment was inserted into pPICZαA and pGAPZαA (Invitrogen, San Diego, Calif.) at the EcoRI and XbaI sites in frame with alpha factor lead sequence in the vector to construct expression vectors pPICTFP and pGAPTFP, containing promoters PAOX1(alcohol oxidase) and PGAP (glyceraldehyde-3-phosphate dehydrogenase), respectively. These constructs were introduced into E. coli JM109, followed by plating onto LB medium containing 25 μg zeocin ml−1 to select positive colonies to prepare DNA for yeast transformation.
Transformation and protein expression
P. pastoris X33 (Invitrogen) was grown in YPD medium (1% yeast extract, 2% peptone and 2% dextrose) and prepared for transformation according to the manufacturer’s instructions. Plasmid DNA (10 μg) was linearized with PmeI (pPICTFP) or BamHI (pGAPTFP) and introduced into P. pastoris X33 by electroporation using an ECM 600 Electro Cell Manipulator (1.5 kV, 129 Ω, 4.9 ms; Gentronics, BTX Instrument Division, San Diego, Calif.). The transformed cells were plated on YPD-zeocin agar (100 μg zeocin ml−1) and incubated at 30°C for 3 days. For constitutive expression (pGAPTFP), single colonies of the transformants were inoculated into YPD medium and incubated at 30°C for 2 days for protease expression. Protease activity in the culture supernatants was measured to select the transformant with the highest protease expression. For inducible expression (pPICTFP), the transformants were inoculated into 8 ml buffered medium with 0.5% glycerol (BMGY), grown for 36 h, pelleted (3,500 g, 10 min), and then re-suspended in 8 ml buffered medium with 0.7% (v/v) methanol (BMMY) to induce protein expression. The induction was repeated every 24 h by adding 0.7% (v/v) methanol to the medium.
Protease purification and activity assay
The expressed r-TfpA enzyme in the culture supernatant of Pichia transformed with pPICTFP was concentrated by ultrafiltration (Millipore, Bedford, Mass.) with a membrane having an apparent molecular cutoff of 10,000 Da. The concentrated protease solution was loaded onto a carboxymethyl (CM) cellulose column (Sigma, St. Louis, Mo.) equilibrated with 10 mM MES [2-(N-morpholino) ethanesulphonic acid], pH 6.0. The bound protein was eluted using a linear gradient from 0 to 0.3 M NaCl in 10 mM MES, pH 6.0 at a flow rate of 0.2 ml/min. Three fractions exhibiting the highest activities were pooled and concentrated using a spin column concentration unit (Millipore). The concentrated protease was loaded onto a Sephadex G-75 gel exclusion column (Sigma) equilibrated with 10 mM MES buffer containing 0.1 M NaCl, pH 6.0. Active protease was eluted as a single peak and used for further analysis. Protease activity was measured in 0.2 M Tris-HCl buffer, pH 8.0 and at 65°C using 1% azocasein (Sigma) as the substrate (Porres et al. 2002). One protease unit (U) was defined as a 0.01 increase in the A440 after 30 min at pH 8 and 65°C. The total protein concentration in the samples was determined by the method of Lowry et al. (1951).
Hydrolysis of various protein substrates
Activity of the expressed protease in hydrolyzing casein, bovine serum albumin (BSA), collagen or keratin was determined by measuring the A280 of the acid-soluble protein hydrolyzate (Makagawa 1975) and a colorimetric method modified from that of Shinmyo et al. (1968). For the modified colorimetric method, a standard curve was generated using solutions of 0–100 μg tyrosine ml−1. One unit of proteolytic activity was defined as the production of 1 μg tyrosine equivalent per minute in 1 ml enzyme solution.
SDS-PAGE, glycosylation, and protein hydrolysis pattern
Endo Hf (New England Biolabs, Beverly, Mass.) was used to determine N-glycosylation of the r-TfpA protease expressed by Pichia transformed with pPICTFP. The reaction was carried out by incubating 100 μl protease solution (0.2 mg/ml) with 0.3 IU Endo Hf in 0.5 M sodium citrate, pH 5.5, and 1 mM PMSF (phenylmethylsulfonyl fluoride) for 5 h at 37°C. After the reaction, aliquots (12 μl) of the mixture together with 3 μl loading dye were loaded in each well of a 12% polyacrylamide gel and subjected to SDS-PAGE using a Mini-Protean II Cell (Bio-Rad, Hercules, Calif.) (Laemmli 1970). Proteins were stained with Coomassie Brilliant Blue R-250 and the molecular weight estimated using an IS-1000 digital imaging system (Alpha Innotech, San Leandro, Calif.) and pre-stained markers (Bio-Rad).
To determine the peptide hydrolysis pattern with different substrates, r-TfpA produced by Pichia transformed with pPICTFP (25 U) was added to 1 ml 0.1% casein or BSA dissolved in 0.2 M Tris-HCl buffer, pH 8.0. The mixture was incubated at 65°C for 0, 15 or 90 min. At each time-point, 100 μl aliquots were taken and transferred to a 1.5-ml Eppendorf tube containing an equal amount of 2 mM PMSF on ice to stop the reaction. After addition of 5× SDS loading buffer and boiling in a water bath for 3 min, the mixture was loaded onto SDS-PAGE (12% gel) to compare protein degradation patterns.
Biochemical characterization
In all the biochemical assays, purified protease from Pichia transformed with pPICTFP was diluted with selected buffers to 10 U ml−1 for optimal temperature assays and to 20 U ml−1 for thermostability and optimal pH assays. Azocasein, the substrate used in all these assays, was dissolved in each selected buffer to a concentration of 1% (w/v). Hydrolysis reactions were started by mixing an equal volume of diluted enzyme and substrate. The pH profile of r-TfpA was determined at 65°C using a modified universal buffer (0.06 M citric acid, 0.06 M K-phosphate, monobasic, 0.06 M boric acid) for pH 4.5–12 and various individual buffer systems (0.2 M citrate for pH 5–6; 0.2 M Tris-HCl for pH 7–8.5, 0.2 M boric acid-Na borate for pH 8–9, and 0.2 M boric acid-NaOH for pH 9.5–11). The optimal temperature of r-TfpA was determined in 0.2 M Tris, pH 8 and at 37, 45, 55, 65, 75, 85 and 95°C. Thermostability of r-TfpA was determined with 0.2 M Tris-HCl buffer, pH 6.0 and 8.0. The diluted samples were incubated for 15 min at 4, 65, 75, 80 and 85°C. After cooling the samples on ice for 30 min, remaining protease activity was measured as described above.
Statistical analysis
Data were analyzed by SAS, and simple t-test was used to compare mean differences. Significance was set at P<0.05 (n=3–6).
Results
Expression of r-TfpA in P. pastoris
Both pPICTFP and pGAPTFP transformants expressed recombinant protease. However, expression was lower for constitutive expression of pGAPTFP than with inducible expression of pPICTFP. Protease activity in the latter was detected 1 day after induction and increased steadily up to 135 U ml−1 6 days after induction, which corresponded to 41 mg l−1 recombinant protease (Fig. 1). However, protease activity reached a plateau at only 32 U ml−1 in the constitutive system after 2 days of fermentation. In both systems, constructs bearing only the mature protein sequence alone did not produce any activity.

Time-course of recombinant thermostable serine protease (r-TfpA) expression in Pichia pastoris transformed with pPICTFP. Cell density was measured as OD600 and also expressed as dry cell mass. Protease activity was measured using azocasein as substrate in 0.2 M Tris-HCl, pH 8.0 and at 65°C. One unit of protease activity was defined as the amount of enzyme that causes an increase in the A440 of 0.01, relative to the control, after 30 min in the test reaction
Protease purification and specific activity
The recombinant protease from Pichia transformed with pPICTFP was purified as a single protein with a molecular size of approximately 21.7 kDa (Fig. 2). A 218-fold purification was achieved for the enzyme with a final yield of 11.7%. Specific activity of the purified protease was estimated at 3,265 U mg−1 (Table 1). Treating TfpA with Endo Hf did not change its molecular weight (Fig. 2).

Representative gel showing the molecular weight and test for glycosylation of the purified r-TfpA protease expressed in pPICTFP transformants of P. pastoris. M Molecular size markers, G purified protease expressed in P. pastoris, DG protease treated with deglycosylase (endo Hf), H f endo Hf only
Enzymatic properties of r-TfpA
The r-TfpA expressed in P. pastoris had an optimal pH of 8.5 with >50% activity between pH 6 and 10 using substrate prepared in the modified universal buffer. There was<20% activity at pH<5 or pH>12. The pH profile assayed with 0.2 M Tris-HCl buffer (pH 7–8.5) and 0.2 M boric acid-Na borate buffer (pH 8–9) showed different optimal pH values: 7–7.5 and 8.5–9, respectively. The expressed r-TfpA had optimal activity at a temperature of 80°C. The relative activity rose in an almost linear fashion from 35 to 80°C, but decreased abruptly once the temperature exceeded 85°C, with only 65% activity remaining at 95°C. Treating the enzyme at 65, 75, or 80°C for 15 min at pH 6 or 8 resulted in no more than 20% activity losses, whereas heating the enzyme at 85°C for 15 min at pH 6 and 8 caused 35 and 44% activity loss, respectively. The recombinant protease showed different activities in hydrolyzing casein, BSA, keratin, and collagen (Fig. 3). Proteolytic activity towards casein was 50–80% higher (P<0.05) than for other substrates. In a time course of protein hydrolysis analyzed by SDS-PAGE (data not shown), all casein was hydrolyzed by r-TfpA into a small band of approximately 7 kDa within 15 min, while BSA had a full spectrum of incompletely digested bands with the same treatment.

Proteolytic hydrolysis of different substrates by purified r-TfpA protease expressed in pPICTFP transformants of P. pastoris. Each substrate (1%) was dissolved or suspended in 0.2 M Tris-HCl buffer, pH 8.0, mixed with an equal volume (200 μl each) of enzyme solution, and incubated at 65°C for 30 min. Bars are means±SD of four replicate samples. Means with different letters were significantly different (P<0.05)
Discussion
It is clear from the present study that both P. pastoris systems can be used to express the protease from T. fusca YX. The inducible system with the AOX1 promoter gave a higher yield than the constitutive system with the GAP promoter. The yield of protease obtained from P. pastoris transformants in the present study is higher than that reported by Gusek and Kinsella (1987) and Lao and Wilson (1996) using T. fusca YX and S. lividans, respectively, as expression hosts. We have also confirmed that the pro-protein region of the enzyme is necessary for functional expression, because only Pichia transformants with constructs carrying the whole sequence, but not those with the mature protein region alone, resulted in activity. This is consistent with other reports stating that most proteases generally require the pro-protein sequences for proper protein folding and for protection from proteolytic hydrolysis in the expression hosts (Kluskens et al. 2002; Porres et al. 2002).
Although Pichia has been increasingly used to manufacture heterologous proteins, the expression yield of T. fusca protease in Pichia in the present study may still be low for practical applications (Noronha et al. 2002). Up to 2–3 g l−1 recombinant protein or enzyme have been expressed extracellularly, and up to 12 g l−1 intracellularly, in the Pichia inducible system (Tschopp et al. 1987; Clare et al. 1991). However, in the present study, r-TfpA was produced with a yield of only 41 mg l−1. The relatively low expression rate of TfpA might be related to: (1) poor protein stability, (2) toxicity of the expressed protein to the host, (3) N-glycosylation related to secretion, and (4) poor codon usage affecting translation efficiency and accuracy. Modifying amino acids at the proteolytic cleavage site (Jang et al. 2001; Demidyuk et al. 2003) may reduce autolytic activity of the expressed protease and improve its stability. Glycosylation of the recombinant protein in Pichia systems is important for efficient protein secretion (Sagt et al. 2000). Although no glycosylation occurred in the mature protein as deglycosylation of r-TfpA resulted in no molecular size change, there are two potential N-glycosylation sites in the pro-protein region of TfpA. It will be useful to find out if introducing new potential glycosylation sites in that region or other regions of the sequence could improve r-TpfA expression and secretion. Optimizing the codon usage of the tpfA sequence may also improve its expression in Pichia as the tfpA sequence contains codons that are rarely, or less frequently, used in Pichia (Kane 1995).
The biochemical properties of r-TfpA expressed by P. pastoris are similar to those from T. fusca and S. lividans, with alkaline optimal pH, high heat stability, and a wide range of protein substrates (Gusek and Kinsella 1987; Kristjansson and Kinsella 1990; Lao and Wilson 1996). The apparent molecular weight (21.7 kDa) of this recombinant protease was different from those (14.5 and 18.1 kDa) reported by Gusek and Kinsella (1987) and Lao and Wilson (1996). This might be due to the different expression host systems causing the pro-protease to be cleaved for activation at different sites by intrinsic protease. This difference may be further confirmed and characterized by N-terminal amino acid sequencing or mass spectrophotometry of r-TfpA expressed in these hosts. The capacity of r-TfpA to hydrolyze various proteins, in particular keratin and collagen, offers a great potential for its application in nutrition, food processing, environmental protection, and the cosmetics industry. If the hydrolysis efficiency of keratin is improved, r-TfpA can be used to convert poorly digestible feather meal into soluble amino acids to meet the nutrient needs of food animals (Moritz and Latshaw 2001; William et al. 1991). This will help prevent the large amount of feather meal produced by poultry production (William et al. 1991) from becoming an environmental hazard.
References
Anwar A, Saleemuddin M (1998) Alkaline proteases: a review. Bioresour Technol 64:175–183
Clare JJ, Romanos MA, Rayment FB, Rowedder JE, Smith MA, Payne MM, Sreekrishna K, Henwood CA (1991) Production of epidermal growth factor in yeast: high-level secretion using Pichia pastoris strains containing multiple gene copies. Gene 105:205–212
Demidyuk IV, Zabolotskaya MV, Safina DR, Kostrov SV (2003) Molecular mechanisms of stabilization of proteolytic enzymes: a model of thermolysin-like microbial metalloproteases. Russ J Bioorg Chem 29:418–425
Gusek TW, Kinsella JE (1987) Purification and characterization of the heat-stable serine proteinase from Thermomonospora fusca YX. Appl Microbiol Biotechnol 128:80–84
Higgins DR, Cregg JM (1988) Introduction to Pichia pastoris. In: Higgins DR, Cregg JM (eds) Pichia protocols. Methods in Molecular Biology series, vol 103. Humana, New York
Jang JW, Ko JH, Kim EK, Jang WH, Kang JH, Yoo OJ (2001) Enhanced thermal stability of an alkaline protease, AprP, isolated from a Pseudomonas sp. by mutation at an autoproteolysis site, Ser-331. Biotechnol Appl Biochem 34:81–84
Kane JF (1995) Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Opin Biotechnol 6:494–500
Kluskens LD, Voorhorst WGB, Siezen RJ, Schwerdtfeger RM, van der Oost GAJ, de Vos WM (2002) Molecular characterization of fervidolysin, a subtilisin-like serine protease from the thermophilic bacterium Fervidobacterium pennivorans. Extremophiles 6:185–194
Kristjansson MM, Kinsella JE (1990) Heat stable proteinase from Thermomonospora fusca. Int J Pept Protein Res 36:201–207
Kumar CG, Takagi H (1999) Microbial alkaline proteases: from a bioindustrial viewpoint. Biotechnol Adv 17:561–594
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lao G, Wilson DB (1996) Cloning, sequencing, and expression of a Thermomonospora fusca protease gene in Streptomyces lividans. Appl Environ Microbiol 62:4256–4259
Layman PL (1986) Industrial enzymes: battling to remain specialties. Chem Eng News 64:11–14
Lowry OH, Rosebrough NJ, Farr AR, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Makagawa Y (1975) Alkaline protease from Aspergillus. Methods Enzymol 41:581–685
Moritz JS, Latshaw JD (2001) Indicators of nutritional value of hydrolyzed feather meal. Poultry Sci 80:79–86
Noronha EF, de Lima BD, de Sa CM, Felix CR (2002) Heterologous production of Aspergillus fumigatus keratinase in Pichia pastoris. World J Microbiol Biotechnol 18:563–568
Porres JM, Benito MJ, Lei XL (2002) Functional expression of keratinase (kerA) gene from Bacillus licheniformis in Pichia pastoris. Biotechnol Lett 24:631–636
Rodriguez E, Mullaney EJ, Lei XG (2000) Expression of the Aspergillus fumigatus phytase gene in Pichia pastoris and characterization of the recombinant enzyme. Biochem Biophys Res Commun 268:373–378
Sagt CMJ, Kleizen B, Verwaal R, de Jong MDM, Muller WH, Smits A, Visser C, Boonstra J, Verkleij AJ, Verrips CT (2000) Introduction of an N-glycosylation site increases secretion of heterologous proteins in yeasts. Appl Environ Microbiol 66:4940–4944
Shinmyo A, Okazaki M, Terui G (1968) Kinetic studies on enzyme production by microbes. IV. Some physiological basis for kinetic studies on acid protease production by Aspergillus niger. J Ferment Technol 46:733–742
Tschopp JF, Sverlow G, Kosson R, Craig W, Grinna L (1987) High level secretion of glycosylated invertase in the methylotrophic yeast Pichia pastoris. Biotechnology 5:1305–1308
William CM, Lee CG, Garlich JD, Shih JCH (1991) Evaluation of a bacterial fermentation product, feather-lysate, as a feed protein. Poultry Sci 70:85–94
Wilson DB (1988) Cellulases of Thermomonospora fusca. Methods Enzymol 160:314–323
Acknowledgements
We would like to thank Dr. David Wilson for providing the gene of tfpA and Diana Irwin for technical help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kim, T., Lei, X.G. Expression and characterization of a thermostable serine protease (TfpA) from Thermomonospora fusca YX in Pichia pastoris. Appl Microbiol Biotechnol 68, 355–359 (2005). https://doi.org/10.1007/s00253-005-1911-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-1911-8