
Abstract
Fruit body initials of Agaricus bisporus contain high levels of urea, which decrease in the following developmental stages until stage 4 (harvest) when urea levels increase again. At storage, the high urea content may affect the quality of the mushroom, i.e. by the formation of ammonia from urea through the action of urease (EC 3.5.1.5). Despite the abundance of urea in the edible mushroom A. bisporus, little is known about its physiological role. The urease gene of A. bisporus and its promoter region were identified and cloned. The coding part of the genomic DNA was interrupted by nine introns as confirmed by cDNA analysis. The first full homobasidiomycete urease protein sequence obtained comprised 838 amino acids (molecular mass 90,694 Da, pI 5.8). An alignment with fungal, plant and bacterial ureases revealed a high conservation. The expression of the urease gene, measured by Northern analyses, was studied both during normal development of fruit bodies and during post-harvest senescence. Expression in normal development was significantly up-regulated in developmental stages 5 and 6. During post-harvest senescence, the expression of urease was mainly observed in the stipe tissue; expression decreased on the first day and remained at a basal level through the remaining sampling period.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Higher fungi of the family Agaricaceae, including Agaricus bisporus, accumulate substantial amounts of urea in their fruit bodies (Hammond 1979; Wagemaker et al. 2005). Fruit body initials of A. bisporus were found to contain high levels of urea, which decrease in the following developmental stages until stage 4 where urea levels increase again, with the exception of stipe tissue where no significant changes were observed. Mushrooms are harvested at stage 4. High amounts of urea have a negative effect on the taste of the mushroom. Furthermore, the ammonia formed when urea is hydrolysed stimulates post-harvest bacterial attack and affects crop quality. Despite the abundance of urea in the edible mushroom A. bisporus, little is known about its physiological role. Urea is chemically inert and soluble to high concentration. Therefore, urea may serve as an osmotically favourable form of fungal nitrogen reserve, especially when mannitol, an osmotically favourable form of carbon and energy reserve stored during active growth, is used as a respiratory substrate in post-harvest senescence (Wood 1985).
Urea formation is a result of purine catabolism and ornithine cycle activity, and urea release accompanies the biodegradation of nitrogenous compounds like arginine, agmatine, allantoin and allantoic acid (Vogels and van der Drift 1976). Two pathways are encountered for urea degradation: hydrolysis via urease (EC 3.5.1.5) or via an ATP-hydrolyzing urease (EC 3.5.1.45). In higher plants, some fungi and many prokaryotes, urea is hydrolyzed by urease. Urease is a nickel-containing enzyme that catalyses urea hydrolysis into ammonia and carbamate. Carbamate is unstable and hydrolyzes spontaneously to carbonic acid and a second ammonia molecule. In other fungi and in green algae, urea is first carboxylated to yield allophanate by an ATP-hydrolyzing urease. The allophanate in turn is hydrolyzed into two molecules each of ammonia and carbon dioxide (Leftley and Syrett 1973; Roon and Levenberg 1972). So far, no data on the presence of a degradation pathway through allophanate in A. bisporus exist. No influence on the ammonia formation was found after addition of ATP to the urease reaction (unpublished data). Urea was shown to be a good nitrogen source for mycelial growth of A. bisporus (Baars et al. 1994), and urease activity was demonstrated in fruit bodies (de Windt et al. 2002). Besides urease, evidence for the presence of a urease inhibitor in cell-free extracts has been presented (Reinbothe et al. 1967; de Windt et al. 2002). Urease allows micro-organisms to use externally and internally generated urea as a source of nitrogen (Mobley and Hausinger 1989).
Expression of a catalytically active urease in bacteria is usually directed by at least seven genes, which in general are arranged in operons (Collins and Dorazio 1993; Koper et al. 2004). Activation of the urease apoenzyme by incorporation of nickel ions into the multimeric molecule is best understood in the bacterium Klebsiella aerogenes (Hausinger et al. 2001). It is accomplished by the coordinated action of four accessory proteins, encoded by ureD, ureE, ureF and ureG. In bacteria, three genes (ureA, ureB and ureC) encode the structural subunits (γ, β and α, respectively) of urease, which associate in an (αβγ)n stoichiometry to form the urease apoenzyme (Jabri et al. 1995). In plants and fungi, the structural urease protein is encoded by one gene that comprises homologues of the three bacterial genes. In this article, we report the isolation and characterization of the first homobasidiomycete urease gene. The A. bisporus urease gene, promoter region and cDNA were isolated and characterized. Moreover, the correlation between the expression of urease at mRNA level and the urea content of fruit bodies at different developmental stages and in post-harvest senescence is discussed.
Materials and methods
Agaricus bisporus strains and culture conditions
Agaricus bisporus Horst U1 and its homokaryotic parents, strains H39 and H97, were obtained from the collection of the Mushroom Experimental Station, Horst, the Netherlands.
A. bisporus fruit bodies were grown and sampled as described before (Wagemaker et al. 2005). Mushrooms for post-harvest studies were sampled and stored as described by Eastwood et al. (2001).
Escherichia coli strains, recombinants DNA techniques and enzymes
Escherichia coli LE392 (Promega) and E. coli XL1 blue (Stratagene) were used for phage amplification/λ DNA isolation and plasmid transformation/propagation, respectively. Standard DNA manipulations were carried out as described in Sambrook et al. (1989). Restriction enzymes and other enzymes used for DNA manipulations were purchased from MBI Fermentas. Plasmid pUC19 (Yanisch-Perron et al. 1985) was used as cloning vector for genomic DNA (gDNA) fragments. The pGEM-T vector system (Promega) was used for cloning polymerase chain reaction (PCR) products. Synthetic oligonucleotide primers were purchased from Biolegio BV and Isogen Bioscience BV. FlexiPrep Kit (Pharmacia Biotech) was used for plasmid isolation.
Cloning of the Agaricus bisporus gene encoding urease
For amplification of A. bisporus DNA encoding a part of the urease gene, synthetic degenerate inosine-containing deoxyoligonucleotide primers, designed from aligned regions conserved in Filobasidiella neoformans (O13465), Coccidiodes immitis (O14420) and Schizosaccharomyces pombe (O00084), were used in a PCR performed on a Tgradient Thermocycler (Biometra) with strain Horst U1 gDNA as a template. PCR primer 1 (5′-GGI CAY GCI CCI GAY ATY ATY-3′) encodes urease amino acids 547–554 of the S. pombe sequence, and primer 2 (5′-CCA IAR IAC IAR RTC NGC-3′) encodes the antisense codons of amino acids 701–706. A nested PCR was performed on 0.1 μl of the first PCR product mix: primer 3 (5′-TIG AYA TGY TIA TGR TNT GYC A-3′) encodes urease amino acids 583–590 and primer 4 (5′-ATO GCO GGR TTD ATO GTR TAU TT-3′) encodes the antisense codons of amino acids 677–684. The initial denaturation step of 10 min at 95°C was used to add the Taq polymerase enzyme as a hot-start and was followed by 30 cycles of 1 min at 95°C, 1 min at 52°C, 2 min 72°C and a final extension step of 5 min at 72°C. The amplified 301-bp product was cloned into pGEM-T (Promega), and four clones were sequenced. Sequences obtained were 100% identical. The fragment was labelled using [α-32P]dCTP by random oligonucleotide priming (HexaLabel DNA Labeling Kit, MBI Fermentas) and used as a probe for the screening of a λEMBL3 genomic library of strain Horst H39 using conditions described previously (Schaap et al. 1996). Plaques giving a positive hybridization signal were purified, and the urease gene was isolated from the λ DNA by ligation of SalI fragments into pUC19 and subsequent sequence analysis of the subclones.
Isolation, sequencing and characterization of ureA cDNA from a mixed primordial small fruit body λ-ZAP-cDNA library (de Groot et al. 1996) were performed as described before (Wagemaker et al. 2005).
Amino acid sequence analysis
Relevant protein sequences were extracted from the SwissProt or the TrEMBL protein databases. The sequences were aligned with the align utility of the Vector NTI Suite software (Informax) and the alignment was edited with GeneDoc.
Northern analysis
Total RNA was isolated from A. bisporus fruit bodies and analyzed as described before (Wagemaker et al. 2005). The PCR product of forward primer 5′-GGC GAC GAA GTC AAA TTT GG-3′ (ure1f) and reverse primer 5′-TCG TTG AGA GTG TCG GTA TG-3′ (ure2r) was labelled with [32P]CTP and used as a probe (Wagemaker et al. 2005). Northern blots were also probed with an A. bisporus 28S ribosomal DNA fragment as loading control and with probes specific for the genes gdhA, gdhB and glnA encoding enzymes of the primary nitrogen metabolism (Kersten et al. 1997, 1999; Schaap et al. 1996).
To study post-harvest development, mushrooms were sampled and stored, and total RNA was isolated as described by Eastwood et al. (2001). The PCR product of the ure1f and ure2r primers was used as probe through the incorporation of [α-32P]dCTP-labelled molecules via the RediPrime random-labelling system (Amersham International).
Isolation of total genomic DNA, Southern blot analysis
Total DNA from strains H39 and H97 was isolated, digested with various restriction enzymes, separated on 0.8% agarose gels and transferred onto Nytran Super Charge membranes by vacuum blotting. DNA blots were washed at 65°C to a final stringency of 0.1× SSC, 0.1% SDS.
The gDNA and cDNA sequences of the urease gene from A. bisporus have been deposited in the GenBank/EMBL databases under the accession numbers AJ748112 and AJ748113, respectively.
Results
The Agaricus bisporus urease gene
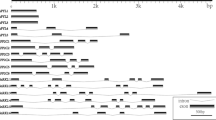
The work on urease was initiated by isolating a gene fragment from A. bisporus strain H39 gDNA by a nested PCR approach using degenerate primers. The degenerated primers were designed from conserved regions in the primary structure of urease sequences from the heterobasidiomycete F. neoformans (Cox et al. 2000) and the ascomycetes C. immitis (Yu et al. 1997) and S. pombe (Tange and Niwa 1997). A nested PCR was performed on the first amplification product, and this resulted in a specific product of 296 bp. Blast search revealed identities of 74 and 75% with F. neoformans and Glycine max urease genes, respectively. The fragment was used as a probe for screening the strain H39 λEMBL3 genomic library. Urease-positive λ DNA was isolated and digested with SalI restriction endonuclease and ligated in pUC19. The isolated promoter region and gene sequence comprised about 3.5 kb distributed over three SalI fragments: clones gU1, gU2 and gU3 (Fig. 1); the full sequence was obtained by the primer walking method. The specific primers were also used in combination with pBluescript vector primers for isolation of the urease cDNA by a PCR on a cDNA library and subsequent sequencing.

Genomic map of the gene ureA encoding the urease of A. bisporus strain H39 based on subcloning of the urease positive lambda phage. gU1, gU2 and gU3 are the subclones of the urease gDNA obtained after digestion with SalI. The gaps in the gDNA sequence indicate the position of introns. For comparison, two bacterial type ureases are included together with the organisation of the apoenzyme
The 3,761 bp gDNA sequence contains an open reading frame (ORF) of 2,514 bp, interrupted by nine introns of 47–71 bp in length. The introns were confirmed by cDNA analysis and all appear to have normal fungal splicing sites (Unkles 1992), except for intron 6, which has gtatt instead of gtnng at its start. The 3′-end of the cDNA was located 50 bp downstream of the TAA stop codon just 2 bp upstream of a putative polyadenylation site (Humphrey and Proudfoot 1988). Seven putative GATA factor binding motifs were found in the 471-bp urease promoter sequence. Furthermore, a putative TATA box, a GC box (box) and a CAAT box involved in transcription initiation could be identified.
Southern analysis of gDNA of strains U1 and its homokaryotic constituents H39 and H97, using a PCR product of primers ure1f and ure2r as a probe, indicated that a single copy of this gene is present in the genome of A. bisporus (Fig. 2). The Southern blot of the H97 strain shows two hybridization products for the ClaI digest instead of one as seen for H39. This is the result of a sequence polymorphism between the two parental strains of Horst U1 located in the wobble base and thus not resulting in a different amino acid (data not shown).

Southern analysis of gDNA of A. bisporus strains H39 and H97 probed with a urease gene PCR product obtained with primers ure1f and ure2r after digestion with different restriction enzymes
The codon usage of the A. bisporus gene showed a limited bias towards a thymine in the third position of a codon (33.6%), similar to the nicotinamide adenine dinucleotide phosphate (NADP)-dependent glutamate dehydrogenase (GDH) (Schaap et al. 1996) and arginase (Wagemaker et al. 2005), whereas in 55.8% of the codons, a pyrimidine was used in the third position of a codon. This is significantly lower compared to the NADP-dependent GDH gene, the arginase gene and the codon usage reported for other A. bisporus genes by Schaap et al. (1999).
Amino acid sequence analysis
The deduced protein sequence of 838 amino acids has a calculated molecular mass of 90,694 Da with a theoretical pI of 5.8. The encoded protein resembles the γ, β and α subunits of the bacterial ureases fused into one protein (Fig. 1) as described before for other eukaryotic ureases. The A. bisporus urease gene provides the first full homobasidiomycete urease protein sequence. An alignment with fungal, plant and bacterial ureases revealed a high similarity among all representative species included. The alignment was used to create a phylogenetic tree (Fig. 3). For comparison, the homobasidiomycete urease DNA sequences of Coprinus cinereus and Phanerochaete chrysosporium were extracted from the genome data provided by the Joint Genome Institute (JGI; http://genome.jgi-psf.org/) and the Broad Institute http://www.broad.mit.edu/). The translated sequences, included in the alignment and tree, showed high identity (75–77%) with the A. bisporus urease amino acid sequence. High conservation of intron positioning was observed among the homobasidiomycetes. Eight introns were conserved in all three species; one intron was conserved only with C. cinereus and one intron was conserved between C. cinereus and P. chrysosporium. C. cinereus possessed one unique intron. The fungal ureases from F. neoformans (O13465), C. immitis (O14420) and S. pombe (O00084) revealed identities of 63, 60 and 60%, respectively, with the A. bisporus urease. For the plant urease from Canavalia ensiformis (P07374) and the bacterial ureases, 60% and 52–55%, respectively, identity was found.

Phylogenetic tree of representative urease family members. Organisms and their accession numbers are A. bisporus (AJ748113), F. neoformans (O13465), C. immitis (O14420), S. pombe (O00084), C. ensiformis (M65260), Solanum tuberosum (Q93WI8), G. max (Q7XAC5), Arabidopsis thaliana (A96699), Helicobacter pylori (X17079), P. mirabilis (M31834) and K. aerogenes (M36068). Urease protein sequences from P. chrysosporium and C. cinereus were extracted from the genome data provided by the JGI and the Broad Institute, respectively. Bar = 10 substitutions per 10 amino acid residues
Regulation of urease expression
Transcriptional regulation of the urease gene was investigated by determining the mRNA levels in various parts of the A. bisporus fruit body during sporophore formation. Tissue samples from developing fruit bodies were used for RNA isolation. Figure 4a shows that the urease gene in normal growing sporophores has a low basal level of expression, which was up-regulated in stages 5, 6 and 7 for stipe and stage 6 for cap tissue. The expression in stipe tissue was much higher when compared to cap tissue and was still increasing in stage 7. The expression in cap tissue decreased in stage 7.

Northern analyses of genes in A. bisporus Horst U1. a Total RNA isolated from stipe and cap tissues cut from fruit bodies in different stages of development (indicated by the numbers). b Total RNA isolated from whole fruit bodies (stage 4) after post-harvest storage for 0 to 24h. c Total RNA isolated from stipe and cap tissues cut from fruit bodies (stage 4) after post-harvest storage for 0 to 2(days. Ten micrograms of total RNA was size-fractionated and hybridized with a urease (ureA), a NADP-dependent GDH (gdhA), a NAD-dependent GDH (gdhB), a glutamine synthetase (glnA) gene fragment or a 28S rRNA gene fragment
If RNA was isolated from the complete fruit body, high expression was observed at harvest (t=0). Expression of the urease gene in post-harvest senescence revealed a down-regulation in the first 24 h post-harvest (Fig. 4b). The slight increase at 15–18 h is not significant when the signals are corrected for the 28S rRNA loading control. For comparison, the blot was also hybridized with probes specific for the NADP-dependent GDHs (gdhA, gdhB) and the glutamine synthetase (glnA) of A. bisporus (Kersten et al. 1997, 1999; Schaap et al. 1996). These enzymes of the primary nitrogen metabolism showed the same expression pattern (Fig. 4b). Studied in more detail, the post-harvest expression of urease appeared to be mainly situated in stipe tissue (Fig. 4c). Expression decreased on the first day, but after another day a basal expression level was still visible, whereas in cap tissue at harvest, a lower expression was observed decreasing below detection level within one day. For gill tissue, no post-harvest mRNA expression was detected.
Discussion
During fruit body development of A. bisporus, urea levels first decrease, but at the stage of harvest (stage 4), urea levels increase again. Upon storage, the high urea content may affect the quality of the mushroom, i.e. by the formation of ammonia from urea through the action of urease. Since little is known about the physiological role of urea, this paper focusses on the characterization and expression of the urease gene and the correlation with the urea content of fruit bodies at different developmental stages and in post-harvest senescence.
Ureases found in bacteria, fungi and higher plants (Mobley et al. 1995) are highly conserved. In most bacterial species, the urease structural enzyme is encoded by three genes: a large catalytic subunit (α), encoded by the ureC gene, and two smaller subunits (β and γ), which are the products of ureB and ureA, respectively. The three subunits can associate as a heteromultimer with (αβγ)n stoichiometry to form the urease apoenzyme. Whereas the ureases of most bacterial species have been found to possess three distinct subunits, Helicobacter species produce ureases with only two distinct subunits (Clayton et al. 1990). Here the subunits β and γ are encoded by one gene ureaA, which is a fusion of the ureA and ureB genes from the bacterial system. In higher plants and fungi, the enzyme is made up of only one unit comprising the three subunits encoded by a single gene (Takashima et al. 1988). The urease of the fungus A. bisporus presented in this work also is the product of one single gene and shows high similarity to the eukaryotic ureases and less to the bacterial enzymes. A complete description of the secondary structure elements and an indication of which residues are orientated to the interior of the (αβγ) trimer of K. aerogenes urease were presented by Jabri et al. (1995) and Jabri and Karplus (1996). Their alignment of 14 bacterial ureases along with jack bean urease showed 189 residues to be identical. Six residues were involved in nickel ligation and five (\({\text{His}}^{{\alpha 219}}\),\({\text{His}}^{{\alpha 320}}\),\({\text{Gly}}^{{\alpha 277}}\),\({\text{Ala}}^{{\alpha 363}}\) and \({\text{Met}}^{{\alpha 364}}\) of the K. aerogenes urease α subunit) were implicated either by biochemical means or by the structure in substrate binding or catalysis. Also in our alignment of the fungal species P. chrysosporium, C. cinereus, F. neoformans, C. immitis and S. pombe along with plant ureases and three bacterial ureases, those residues are conserved. A total of 178 residues were proposed to be important in hydrogen bonding and hydrophobic interactions within the αβγ unit or in interactions to build the (αβγ)3 trimer; 27 of these residues were not identical in our alignment.
The clear correspondence between the amino acid sequence of the fungal and plant ureases that consist of single units and the two or three subunit bacterial ureases indicates the occurrence of gene fusion or disruption events during evolution (Fig. 3; Hausinger 1993). Sequences that are very similar to the intron splice acceptor consensus sequences were found in the DNA sequence between the ureA and ureB ORFs of Proteus mirabilis. On the basis of this, it was suggested that the origin of ureA and ureB may have resulted from an intron site that is not functional in prokaryotes (Mobley et al. 1995). Such a scenario is consistent with a eukaryote-to-prokaryote urease gene exchange during evolution. However, the absence of conservation of one or more introns among the eukaryotic ureases while they share higher identity than with P. mirabilis or other bacterial urease sequences and the presence of a set of accessory genes in one operon in bacteria make a gene fusion more likely.
The accumulation of urea in the fruit body of A. bisporus was proposed to facilitate the translocation of water and metabolites in fruit bodies (Donker and van As 1999), which is required for the production of spores. Urea could be used as an osmoticum to drive cell expansion and, therefore, cap expansion. Expansion is more important to cap opening than growth. Data suggest that sugars drive expansion during natural development (Wood 1985), but in post-harvest most sugars and soluble proteins are metabolised (Hammond and Nichols 1979). Therefore, urea could be involved in continued cap expansion during post-harvest development. Secondly, in post-harvest development, urea was proposed to be the end product of catabolic pathways (Reinbothe et al. 1967; Foret 1990). The observed difference in urea accumulation between the stipe and the rest of the fruit body (Wagemaker et al. 2005) illustrates the dynamics of the translocation process of water and metabolites. The expression of the urease gene is in accordance with this hypothesis. The expression of urease during normal growth was higher in stipe tissue compared to other tissues and prolonged to stage 7, where also no accumulation of urea was observed (Wagemaker et al. 2005). The expression in cap tissue stopped at stage 6 when the urea content increased. For cap tissue, the higher accumulation of urea in post-harvest senescence when compared to normal development stages 5, 6 and 7 did not correlate with a higher expression of the arginase gene (Wagemaker et al. 2005) but can be explained by an earlier stop in urease expression.
In C. cinereus, high urease activity in mycelium and stipe tissue was demonstrated but no detectable activity in cap tissue was found, which correlated with the accumulation of urea in cap tissues (Ewaze et al. 1978). A causal relationship between urea accumulation and water influx into developing cap tissues was proposed, identical to the situation in A. bisporus. The up-regulation of ammonium-scavenging enzymes, NADP-linked GDH (NADP–GDH) and glutamine synthetase in cap tissue was linked with the start of karyogamy (Moore et al. 1987a). It was suggested that the role of the glutamine synthetase and NADP–GDH ammonium scavenging is to maintain an ammonium-free environment in a cell during sporulation, since such processes are inhibited by the presence of ammonium (Moore et al. 1987b; Pinon 1977). An expression study of gdhA, gdhB and glnA and activity measurements of the corresponding enzymes during sporophore formation and post-harvest development will unravel the scavenging role of the primary ammonia-assimilating enzymes in A. bisporus. A preliminary Northern analysis (Fig. 4b) provides evidence that expression of these genes is correlated with urease expression in this fungus.
The urea decrease in the premature stages of sporophore formation indicates that urea might serve as a nitrogen reserve used for synthesis of protein, cell wall and other nitrogenous compounds needed in the development of the fruit body. The expression of the urease gene presented in this paper is in line with the observed urea levels (Wagemaker et al. 2005) and activities measured (Reinbothe et al. 1967). The decrease in urea content of the growing sporophore was thus not only the result of the dilution effect as the fruit body develops and grows to a larger volume. The urease activity demonstrated indicates that ammonium is produced and can be reassimilated by the primary ammonium assimilation pathways addressed by Baars et al. (1996) and Kersten et al. (1997, 1998, 1999). For mature fruit bodies of A. bisporus, however, no reassimilation of urea–ammonia was determined as was shown for puffballs of Lycoperdon pyriforme (Reinbothe et al. 1967). An increase in alanine and proline concentration after urea administration suggested assimilation of the urea nitrogen. Because urease activity is evident and high levels of urea are present, a different cell localization of the urease enzyme and the high level of urea might be speculated. Sequence data on the arginase (Wagemaker et al. 2005) and urease genes indicate that production of urea as well as the degradation are cytosolic activities, since the encoded protein sequences do not show translocation signals. Identification and localization of the previously reported inhibitor (Reinbothe et al. 1967; de Windt et al. 2002) and its role in controlling the urease enzyme activity will give a better understanding of the various functions that urea accumulation fulfils in A. bisporus fruit body formation. The expression of the urease enzyme as a reporter-fusion protein would enable one to study the enzyme and its localization in more detail.
References
Baars JJP, Op den Camp HJM, Hermans JMH, Mikes V, van der Drift C, Van Griensven LJLD, Vogels GD (1994) Nitrogen assimilating enzymes in the white button mushroom Agaricus bisporus. Microbiology 140:1161–1168
Baars JJP, Op den Camp HJM, van der Drift C, Sonneberg ASM, Van Griensven LJLD (1996) A review of primary nitrogen metabolism in Agaricus bisporus. CMRI Newsl 3:1–15
Clayton CL, Pallen MJ, Kleanthous H, Wren BW, Tabaqchali S (1990) Nucleotide sequence of two genes from Helicobacter pylori encoding for urease subunits. Nucleic Acids Res 18:362–363
Collins CM, Dorazio SEF (1993) Bacterial ureases: structure, regulation of expression and role in pathogenesis. Mol Microbiol 9:907–913
Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR (2000) Urease as a virulence factor in experimental cryptococcosis. Infect Immun 68:443–448
De Groot PWJ, Schaap PJ, Sonnenberg ASM, Visser J, Van Griensven LJLD (1996) The Agaricus bisporus hypA gene encodes a hydrophobin and specifically accumulates in peel tissue of mushroom caps during fruit body development. J Mol Biol 257:1008–1018
de Windt FE, Wagemaker MJM, Op den Camp HJM, van der Drift C (2002) Purine degradation in the edible mushroom Agaricus bisporus. Folia Microbiol 47:672–676
Donker HCW, van As H (1999) Cell water balance of white button mushrooms (Agaricus bisporus) during its post-harvest lifetime studied by quantitative magnetic resonance imaging. Biochim Biophys Acta 1427:287–297
Eastwood DC, Kingsnorth CS, Jones HE, Burton KS (2001) Genes with increased transcript levels following harvest of the sporophore of Agaricus bisporus have multiple physiological roles. Mycol Res 105:1223–1230
Ewaze JO, Moore D, Stewart GR (1978) Co-ordinate regulation of enzymes involved in ornithine metabolism and its relation to sporophore morphogenesis in Coprinus cinereus. J Gen Microbiol 107:343–357
Foret V (1990) Aspects métabolizues de la fructification d’Agaricus bisporus (Lange) Imbach. Cryptogam Mycol 11:255–287
Hammond JBW (1979) Changes in composition of harvested mushrooms (Agaricus bisporus). Phytochemistry 18:415–418
Hammond JBW, Nichols R (1979) Carbohydrate metabolism in Agaricus bisporus: changes in non-structural carbohydrates during periodic fruiting (flushing). New Phytol 83:723–730
Hausinger RP (1993) Biochemistry of nickel. Plenum, New York
Hausinger RP, Colpas GJ, Soriano A (2001) Urease: a paradigm for protein-assisted metallocenter assembly. ASM News 67:78–84
Humphrey T, Proudfoot NJ (1988) A beginning to the biochemistry of polyadenylation. Trends Genet 4:243–245
Jabri E, Karplus PA (1996) Structures of the Klebsiella aerogenes urease apoenzyme and two active-site mutants. Biochemistry 35:10616–10626
Jabri E, Carr MB, Hausinger RP, Karplus PA (1995) The crystal structure of urease from Klebsiella aerogenes. Science 286:998–1004
Kersten MASH, Müller Y, Op den Camp HJM, Vogels GD, Van Griensven LJLD, Visser J, Schaap PJ (1997) Molecular characterization of the glnA gene encoding glutamine synthetase from the edible mushroom Agaricus bisporus. Mol Gen Genet 256:179–186
Kersten MASH, Baars JJP, Op den Camp HJM, Van Griensven LJLD, van der Drift C (1998) Regulation of glutamine synthetase from the white button mushroom Agaricus bisporus. Arch Biochem Biophys 364:228–234
Kersten MASH, Müller Y, Baars JJP, Op den Camp HJM, van der Drift C, Van Griensven LJLD, Visser J, Schaap PJ (1999) NAD+-dependent glutamate dehydrogenase of the edible mushroom Agaricus bisporus: biochemical and molecular characterization. Mol Gen Genet 261:452–462
Koper TE, El-Sheikh AF, Norton JM, Klotz MG (2004) Urease-encoding genes in ammonia-oxidizing bacteria. Appl Environ Microbiol 70:2342–2348
Leftley JW, Syrett PJ (1973) Urease and ATP: urea amidolyase activity in unicellular algae. J Gen Microbiol 77:109–115
Mobley HLT, Hausinger RP (1989) Microbial urease: significance, regulation, and molecular characterization. Microbiol Rev 53:85–108
Mobley HLT, Island MD, Hausinger RP (1995) Molecular biology of microbial ureases. Microbiol Rev 59:451–480
Moore D, Liu M, Kuhad RC (1987a) Karyogamy-dependent enzyme derepression in the basidiomycete Coprinus. Cell Biol Int Rep 11:335–341
Moore D, Horner J, Liu M (1987b) Co-ordinate control of ammonium-scavenging enzymes in fruit body of Coprinus cinereus avoids inhibition of sporulation by ammonium. FEMS Microbiol Lett 44:239–242
Pinon R (1977) Effects of ammonium ions on sporulation of Saccharomyces cerevisiae Exp Cell Res 105:367–378
Reinbothe H, Wasternack C, Miersch J (1967) Harnstoff-Metabolismus by Basidiomyceten: IV. Untersuchungen zur Physiologie des Harnstoffs. Flora 158:27–57
Roon RJ, Levenberg B (1972) Urea amidohydrolase I. Properties of the enzyme from Candida utilis. J Biol Chem 247:4107–4113
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schaap PJ, Müller Y, Baars JJP, Op den Camp HJM, Sonnenberg ASM, Van Griensven LJLD, Visser J (1996) Nucleotide sequence and expression of the gene encoding NADP+-dependent glutamate dehydrogenase (gdhA) from Agaricus bisporus. Mol Gen Genet 250:339–347
Schaap PJ, de Groot PWJ, Müller Y, Sonnenberg ASM, Visser J (1999) Analysis of the structure of housekeeping genes of Agaricus bisporus and their spatial expression in fruit bodies. Ph.D. thesis, University of Nijmegen
Takashima K, Suga T, Mamiya G (1988) The structure of jack bean urease. The complete amino acid sequence, limited proteolysis and reactive cysteine residues. Eur J Biochem 175:151–165
Tange Y, Niwa O (1997) Identification of the ure1+ gene encoding urease in fission yeast. Curr Genet 32:244–246
Unkles SE (1992) Gene organization in industrial filamentous fungi. In: Kinghorn JR, Turner G (eds) Applied molecular genetics of filamentous fungi. Chapman & Hall, London, pp 28–53
Vogels G, van der Drift C (1976) Degradation of purines and pyrimidines by microorganisms. Bacteriol Rev 40:403–468
Wagemaker MJM, Welboren W, van der Drift C, Jetten MSM, Van Griensven LJLD, Op den Camp HJM (2005) The ornithine cycle enzyme arginase from Agaricus bisporus and its role in urea accumulation in fruit bodies. Biochim Biophys Acta 1682:107–115
Wood DA (1985) Production and roles of extracellular enzymes during morphogenesis of basidiomycete fungi. In: Moore DM, Casselton LA, Wood DA, Frankland JC (eds) Developmental biology of higher fungi. Cambridge University Press, Cambridge, MA, pp 375–387
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119
Yu JJ, Smithson SL, Thomas PW, Kirkland TN, Cole GT (1997) Isolation and characterization of the urease gene (URE) from the pathogenic fungus Coccidioides immitis. Gene 198:387–391
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wagemaker, M.J.M., Eastwood, D.C., van der Drift, C. et al. Expression of the urease gene of Agaricus bisporus: a tool for studying fruit body formation and post-harvest development. Appl Microbiol Biotechnol 71, 486–492 (2006). https://doi.org/10.1007/s00253-005-0185-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0185-5