
Abstract
Pseudomonas strain CT14 was isolated from activated sludge. Strain CT14 contained a 55, 216 bp plasmid that was characterized by sequence analysis. The plasmid had a modular structure with 51 open reading frames (ORFs) that were distributed between two clearly demarcated domains. Domain I primarily contained genes for plasmid-related functions and a novel origin of replication. Domain II bore evidence of extensive transposition and recombination. Domain II contained several genes from a meta-cleavage pathway for aromatic rings. These genes appeared to have been recruited from different hosts. This observation suggests that sequencing pCT14 may have revealed an intermediate stage in the evolution of a new assemblage of meta-cleavage pathway genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The degradation genes for many xenobiotic organic compounds have been found on catabolic plasmids and other mobile genetic elements (Nojiri et al. 2004). Several large catabolic plasmids have now been sequenced (e.g., Chattoraj 2000; Greated et al. 2002; Igloi and Brandsch 2003; Li et al. 2004; Maeda et al. 2003; Romine et al. 1999; Vedler et al. 2004). Although these plasmids are diverse and encode a variety of different degradation pathways, all of these plasmids have a modular organization (Thomas 2000). Each module is a cluster of genes encoding related functions, e.g., genes for replication and stable segregation form a distinct cluster, and genes for conjugation form a different distinct cluster. The modular organization of the Tol pathway xyl genes on plasmid pWW0 is typical for plasmid-associated degradation genes (Greated et al. 2002). The xyl genes are organized into two operons. The upper pathway operon encodes enzymes for oxidation of toluene to benzoate. The lower pathway operon encodes enzymes that cleave the aromatic ring and produce intermediates of the TCA cycle. It is most likely that clustering of functionally related genes has evolved to minimize the probability that recombination will disrupt a set of genes that are useful only in the presence of each other (Thomas 2000).
The catabolic plasmids that have been sequenced are generally from bacteria that were originally isolated from soil. However, because the selective conditions in wastewater bioreactors are different than in soil and other natural environments, the microbial diversity in wastewater bioreactors provides a framework in which new biochemical pathways can evolve (Bramucci and Nagarajan 2000). One factor influencing natural selection in industrial wastewater bioreactors, particularly those that recycle biomass as part of the activated sludge process, is higher concentrations of metabolically active bacteria than are found in many natural environments (Bitton 1994). Accordingly, the unique growth conditions in wastewater bioreactors should facilitate formation of novel biochemical pathways through horizontal gene transfer and recruitment of different genes from diverse hosts into a single host. Indeed, bacteria with novel genes and biochemical pathways have been isolated from industrial wastewater bioreactors (Bramucci et al. 2002a,b; Cheng et al. 2000; Kostichka et al. 2001).
In this report, we describe the sequence of a plasmid from a Pseudomonas strain isolated from activated sludge. The plasmid had a modular structure and a novel origin of replication. One region of the plasmid bore evidence of extensive transposition and recombination. This same region contained an incomplete meta-cleavage operon for aromatic rings. Inasmuch as the meta-cleavage operon had genes that were apparently derived from different sources, it is possible that the plasmid represents an intermediate stage in the formation of a new assemblage of meta-cleavage pathway genes.
Materials and methods
Isolation of strain CT14
Liquid S12 medium, S12 agar, Luria–Bertani (LB) medium, and LB agar were used for culturing bacteria (Bramucci et al. 2002a; Sambrook et al. 1989). S12 liquid medium was supplemented with toluene by adding the toluene directly to the culture medium. S12 agar plates were supplemented with toluene by placing 20 μl of toluene on the interior of each petri dish lid. The petri dishes were sealed with parafilm and incubated upside down at 25°C.
Activated sludge from an industrial wastewater bioreactor was inoculated into 10 ml of S12 medium with 100 mg/l toluene in a 125-ml screw-cap Erlenmeyer flask. The enrichment culture was incubated at 25°C with reciprocal shaking. The enrichment culture was maintained by adding 100 mg/l of toluene every 2–3 days. The culture was diluted 1:10 every 10 days. Bacteria that utilize toluene as a sole source of carbon and energy were isolated at 25°C by spreading samples of the enrichment culture onto S12 agar supplemented with toluene. Representative bacterial colonies were tested for the ability to use toluene as a sole source of carbon and energy on S12 agar.
The 16S rRNA gene sequence of strain CT14 was amplified by the polymerase chain reaction with primers HK12 and HK13 and sequenced essentially as previously described (Bramucci et al. 2002a). The 16S rRNA gene sequence was used as the query sequence for a BLAST search of GenBank for similar sequences (Altschul et al. 1997). Isolate CT14 was deposited with the American Type Culture Collection (Manassas, VA, USA) and assigned accession number PTA-4995.
Isolation of plasmid DNA
Strain CT14 was inoculated into 100 ml of LB medium and incubated for 17 h at 37°C with reciprocal shaking. The cells were collected by centrifugation (10 min, 2,300×g, 4°C), resuspended in 8 ml of Solution 1 (50 mM Tris–HCl, pH 7.5, 10 mM EDTA, 100 ug/ml RNase A), and lysed by adding 8 ml of Solution 2 (0.2 M NaOH, 1% SDS). The cleared lysate was neutralized by adding 8 ml of Solution 3 (1.32 M potassium acetate, pH 4.8) and gently inverting until a white precipitate formed. The supernatant was collected by centrifugation (30 min, 13,000×g, 4°C) and transferred to new tubes. The DNA was precipitated by adding 4M NaCl (final concentration 0.4 M) and 2.5 vol of 100% ethanol and placing the tubes at −20°C for 17 h. The precipitated DNA was recovered by centrifugation (45 min, 13,000×g, 4°C). The white pellet was washed with cold 70% ethanol, dried by rotary evaporation, dissolved in 250 μl of deionized water, and stored at −20°C.
Plasmid DNA was purified by field inversion gel electrophoresis (FIGE) using a Hoeffer Scientific Instruments PS500XT DC power supply and PC500 SwitchBack Pulse Controller. Total genomic DNA was electrophoresed through 1.0% low melting temperature agarose (SeaPlaque) in 0.5× TBE buffer at 6 to 9 V/cm with a 10-min run-in, 50–80 s pulse time, and 3:1 For/Rev switch time for 24 to 28 h (Sambrook et al. 1989). The eletrophoretic gels were stained in 0.5× TBE buffer containing ethidium bromide (1 ug/ml). The DNA bands that corresponded to pCT14 were excised and placed into microfuge tubes. The gel slices were melted at 70°C for 5 min and extracted with an equal volume of tris-saturated phenol. The samples were centrifuged in a microfuge at the highest speed for 10 min The aqueous layer was transferred to a new microfuge tube and extracted twice with 1:1 chloroform/phenol (EM Science) and 24:1 chloroform/isoamyl alcohol (EM Science). After the final extraction, the aqueous layer was adjusted to 0.4M NaCl final concentration. The DNA was precipitated with 2 vol of ethanol at −70°C overnight. The precipitated DNA was centrifuged in a microfuge at the highest speed for 15 min, and the pellet was washed with 70% ethanol. The pellet was dried and resuspended in 50 μl of sterile deionized water.
Sequencing of pCT14
Plasmid DNA was diluted to a final volume of 1.5 ml and placed in a modified Aeromist nebulizer. The modifications consisted of disassembling the nebulizer and removing the outer lip of the inner cone and then replacing the cone upside-down within the nebulizer. The nebulizer was then reassembled. The plasmid DNA was transferred to the nebulizer. Using the included tubing, the nebulizer was attached to a filtered compressed air line with a pressure regulator set for 18 psi. Air was blown through the nebulizer for 30 s while tapping the nebulizer on the tabletop to dislodge any droplets attached to the sides of the nebulizer. Five hundred microliters of the DNA sample was removed, and compressed air was reapplied for an additional 15 s. Another 500 μl of the sample was removed, and compressed air was reapplied for another 15 s. The three samples were then recombined and run on a 1% 1X TEA low melting temperature gel. A section of the gel corresponding to 2 to 4 kb based was excised from the gel and purified using a commercial kit (Qiagen catalog #). The fragments were polished with T4 DNA polymerase ligated into SmaI digested pUC18 (Ready-to-Go, Pharmacia-Biotech) at 22°C for 16 h. The ligated DNA was transformed into DH5-α Maximum Efficiency Competent cells (Gibco-BRL). Cells were plated on Q-plates containing LB+Ampicillin (50 μg/ml)+X-gal. Plates were placed at 37°C overnight. White colonies containing inserts were picked using a Q-bot colony picker and inoculated into 96 or 384 well microtiter plates for high-throughput sequencing.
Plasmid sequence assembly
Sequence data obtained from the high-throughput sequencing facility were assembled using Sequencher (GeneCodes) and Phred/Phrap. Assembled data were manually edited and annotated. All potential open reading frames (ORFs) were compared against the GenBank database using BLASTX and BLASTP (Altschul et al. 1997). The complete pCT14 nucleotide sequence was deposited in GenBank (Accession number DQ126685).
Results
Isolation and characterization of strain CT14
Strain CT14 was isolated from a toluene enrichment culture. This isolate utilized toluene, m-xylene, and the corresponding intermediates of the Tol pathway as sole sources of carbon and energy (data not shown). Microscopic examination revealed that strain CT14 was a Gram-negative rod. The DNA encoding 16S rRNA in strain CT14 was amplified by PCR, sequenced, and compared to 16S rRNA gene sequences contained in GenBank. The CT14 16S rRNA gene sequence had 100% similarity to Pseudomonas veronii (GenBank Accession number AB056120).
An agarose gel electrophoresis procedure that involved in situ lysis of bacteria was used to initially test strain CT14 for plasmid DNA (Eckhardt 1978). A single plasmid was detected (data not shown). The plasmid was sequenced, and the BlastX algorithm was used to assign putative functions to ORFs by comparison to sequences in GenBank.
pCT14
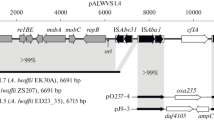
Plasmid pCT14 was 55,216 bp in size and had at least 51 ORFs that were divided between two clearly demarcated domains (Fig. 1). Most of the ORFs in Domain I (1–26,829) were predicted to be genes for plasmid replication and stability (Table 1). Domain II (26,830–55,216) contained a set of genes encoding part of a meta-cleavage pathway for chlorinated aromatic rings.

Map of pCT14 genetic features identified by sequence analysis. The broken line separates Domain I from Domain II. Predicted functions of proteins as indicated by color: pink, plasmid replication, stability, and conjugation; green, transposition and site-specific recombination; blue, mercury resistance; magenta, meta-cleavage pathway enzymes; yellow, conserved hypothetical proteins and other functions. Other features: light blue, long direct repeats (LDR), inverted repeats (IR); red, oriV and oriT
Most of the putative translation products derived from the ORFs in Domain I were similar to polypeptides encoded by various broad host range plasmids. However, pCT14 did not have extensive nucleotide base sequence homology to any characterized plasmid except for regions associated with transposable elements. The interaction of catabolic transposons and mobilizable plasmids is a key factor in the distribution of degradation genes among bacteria exposed to xenobiotic compounds (Nojiri et al. 2004; Top and Springael 2003). Comparison of pCT14 with several different catabolic plasmids indicated that pCT14 had more transposase genes relative to its size than other plasmids (Table 2). This observation suggests that pCT14 has passed through one or more environments that were dynamic for transposition.
pCT14 replication and partitioning functions
The putative replication region was contained within Domain I and was similar to the replication regions of iteron-containing plasmids. The replication regions of iteron-containing plasmids typically have several 20-bp direct repeats (iterons), inverted repeats and DnaA boxes that form the ori and are located immediately upstream from a rep gene (Chattoraj 2000). The iterons bind the cognate Rep protein so that DnaA can initiate melting within ori. In addition to being necessary for initiation of replication, the inverted repeats and iterons act as operators for the rep gene. Although the distinguishing sequence features of most iteron-containing plasmids are located upstream of the rep gene, IncHI plasmids and IncQ differ. The IncHI2 plasmid R478 has two different iteron-regulated replicons (Gardner et al. 2001). Iterons are located upstream and downstream of the repA genes for both replicons. The iterons of the IncQ-like plasmid pTC-F14 are downstream from a repABC operon (Page et al. 2001).
The pCT14 rep gene was adjacent to a series of four direct 20-bp repeats, two 20-bp inverted repeats and two putative DnaA boxes (Fig. 2). One of the DnaA boxes overlapped one of the inverted repeats. The pCT14 ori region differed from typical iteron-containing plasmids in that the direct repeats, inverted repeats, and DnaA boxes comprising the ori region were downstream of the rep gene. No interon-like sequences or long inverted repeats were located upstream of the pCT14 rep gene as is the case for both replicons associated with R478 (Gardner et al. 2001). In addition, the genes encoding putative replication and partitioning functions for pCT14 were not in a single operon as on pTC-F14 and other repABC replicons (Fig. 1; Venkova-Canova et al. 2004). Hence, control of replication for pCT14 is most likely not the same as the IncHI and IncQ plasmids.

Map of the pCT14 oriV region. Sequence colors: pink, rep; green, tnpA2; light blue, direct repeats and inverted repeats; orange, DnaA boxes
The rep gene was separated from putative stability genes by a presumptive transposon. The presumptive transposon contained putative genes for a transposase (tnpA2) and a recombinase (tnpR) that were located between inverted 26-bp repeats. The presumptive transposon was flanked by two 6-bp direct repeats (...GATACT...), suggesting the possibility that a target site had been duplicated during transposition.
Plasmid pCT14 carried two sets of ORFs that were similar to genes associated with plasmid stability. Neither set of genes has been extensively studied. The vagC and vagD genes were originally recognized on the basis of plasmid mutations that decreased virulence of Salmonella dublin (Pullinger and Lax 1992). Genes that are closely related to vagC and vagD have been implicated in plasmid maintenance (Radnedge et al. 1997). Mutations in stb genes have been demonstrated to cause increased rates of plasmid loss (Paterson et al. 1999). Although homologs of both systems have been observed to be located on a single plasmid such as Salmonella typhimurium plasmid R64 (GenBank accession number NC_005014), this occurrence seems to be rare.
Plasmid mobilization functions
Domain I contained two putative conjugation genes. However, pCT14 lacked most of the genes that would be necessary for self-mobilization. The sequence upstream from traJ contained a possible origin for DNA transfer (oriT). The putative oriT was located in an A/T rich region and consisted of a 29-bp sequence that included the nic site of plasmid pMUR274 (Paterson and Iyer 1997).
Genes for meta-cleavage of aromatic rings
A portion of Domain II was essentially the same as a fragment of DNA that was previously cloned from Pseudomonas putida GJ31 (Mars et al. 1999). The cloned GJ31 DNA includes four genes that encode part of a meta-cleavage pathway: cbzT (ferredoxin), cbzE (chlorocatechol 2,3-dioxygenase), cbzX (unknown function), and the 5′ end of cbzG (2-hydroxymuconic semialdehyde dehydrogenase). These genes have been found in several bacterial isolates that degrade chlorocatechol via a meta-cleavage pathway (Goebel et al. 2004). The pCT14 sequence included the entire cbzG gene. In addition, two ORFs were located downstream of cbzG. The first ORF (bphK) encoded a putative glutathione S-transferase that may be involved in dehalogenation of intermediates from degradation of polychlorinated biphenyls (Hofer et al. 1994). The second ORF (tdnG) encoded another meta-cleavage pathway enzyme, 2-oxopent-4-dienoate hydratase.
If the pCT14 meta-cleavage pathway genes enable strain CT14 to degrade chlorinated aromatics, then strain CT14 should grow with at least one of the isomers of chlorobenzoic acid as a source of carbon and energy. Although strain CT14 readily utilized benzoic acid as a sole source of carbon and energy, strain CT14 failed to do so with the isomers of chlorobenzoic acid (data not shown).
All of the meta-cleavage pathway genes except tdnG were flanked by two long direct repeats (LDR1 and LDR2) that were each 1693 bp in length (Fig. 1). Both repeats had a single ORF that corresponded to the 3′ portion of a transposase gene. The LDR2 ORF extended in the 5′ direction beyond the upstream border of LDR2, resulting in a complete ORF (tnpA1) whose deduced translation product was similar to an IS801-like transposase encoded by pADP-1 (Martinez et al. 2001). The tdnG gene was located between tnpA1′ (the 3′ portion of tnpA1 that was located on LDR1) and tnpA5, which was the 5′ portion of another transposase gene. The deduced tnpA5 transcription product had 100% identity to the corresponding portion of an IS1071-like transposase (GenBank accession number AAK50306), indicating that tnpA1′ and tnpA5 were unlikely to be parts of the same gene. Differences in base composition as reflected by differences in % G+C are commonly accepted as indications of different origins for genes (Lawrence and Roth 1996; Ochman and Lawrence 1996). Such differences are frequently used to argue for exogenous origin of a gene or set of genes (Dogra et al. 2004). The difference in % G+C for tnpA1′ (66.2% G+C) and tnpA5 (56.5% G+C) supported the conclusion that these sequences were derived from different genes.
The position of tdnG provided a strong indication that tdnG had been recruited onto pCT14 separately from the other meta-cleavage pathway genes. The tdnG sequence was located between two apparently unrelated transposase sequences (i.e., tnpA1′ and tnpA5). Furthermore, tdnG was separated by LDR1 from the other meta-cleavage pathway genes that were clustered between LDR1 and LDR2 (Fig. 1). The G+C content for tdnG was 71.4%, whereas the other meta-cleavage pathway genes on pCT14 ranged from 55.8 to 62.3% (Table 3). Hence, the differences in % G+C values for tdnG and the other meta-cleavage pathway genes supported the conclusion that tdnG had been recruited onto pCT14 separately from the other meta-cleavage pathway genes.
Tn5053
The two inverted copies of mercury resistance transposon Tn5053 that flanked the ends of Domain II formed an interesting feature of pCT14 (Kholodii et al. 1993, 1995). Tn5053 displays a marked preference for inserting into the res regions associated with site-specific recombinases of the resolvase/DNA invertase family (Leschziner et al. 1995; Minakhina et al. 1999). Independent Tn5053 insertions usually have the same orientation, i.e., the merR end of the transposon is adjacent to the site-specific recombinase of the target element. Because insertion of Tn5053 involves duplication of the target site, the transposon is flanked by 5-bp direct repeats. If Domain II functioned as a Tn5053-based transposable element, then Domain II should be flanked by 5-bp direct repeats that represent duplication of a single insertion site. Otherwise, the two copies of Tn5053 should have different sets of 5-bp direct repeats that represent duplication of independent target sites.
Tn5053-I was oriented on pCT14 so that merR was adjacent to an ORF whose putative translation product was identical to the Tn5501 resolvase (Lauf et al. 1998). The region containing Tn5053-I was flanked by 38-bp inverted repeats that were identical to the terminal inverted repeats of cryptic Tn3-family transposon Tn5501 (Lauf et al. 1998). Deleting the Tn5053-I sequence from the pCT14 sequence resulted in the remaining nucleotide sequence between the two inverted repeats being 97% identical to Tn5501. Hence, Tn5053-I had inserted into a transposon that was closely related to Tn5501. The Tn5053-I insertion site on Tn5501 was marked 4-bp direct repeats (...TACC...).
Tn5053-II was oriented so that merR was adjacent to an ORF whose translation product was similar to putative resolvases and invertases (res; 109–726). The insertion site of Tn5053-II was between res and an ORF whose translation product was similar to the IS801-like transposase encoded by pADP-1 (Martinez et al. 2001). Although Tn5053-II was associated with an external transposase and resolvase that presumably recruited Tn5053-II into pCT14, inverted repeats that defined the ends of a corresponding transposable element could not be identified. The Tn5053-II insertion site was marked by 4-bp direct repeats (...CAGG...) that differed from Tn5053-I. These observations indicated that Tn5053-II most likely inserted into pCT14 independently of Tn5053-I.
Discussion
Inasmuch as the xyl genes for degradation of toluene are present on catabolic plasmids in 85–100% of the bacterial strains that express the Tol pathway, it was expected that pCT14 would encode toluene degradation in strain CT14 (Sentchilo et al. 2000; Williams and Worsey 1976). Although no xyl genes were present on pCT14, the plasmid contained several meta-cleavage pathway genes that have been implicated in degradation of chloroaromatics. The role of the pCT14 meta-cleavage pathway in strain CT14 is unclear because strain CT14 did not grow on chlorobenzoates. Nevertheless, these genes are interesting because they are located on a portion of the plasmid that has undergone several transpositions and possibly other recombination events.
It is tempting to speculate that Domain II transposed into pCT14 as a single complex element because the two copies of Tn5053 and the corresponding target elements flanked the meta-cleavage pathway genes. This scenario is unlikely because the Tn5053 transpositions apparently occurred independently of each other. The presence of Tn5501, the tnpA1/res element, and portions of two additional transposases indicated that other transposition events have occurred during the formation of Domain II. At least four transpositions have shaped the complex structure of Domain II, i.e., two insertions of Tn5053 and insertions for the two elements that recruited Tn5053. It was evident that insertion of Tn5501 proceeded insertion of Tn5053-I. Similarly, insertion of the tnpA1/res element proceeded insertion of Tn5053-II. However, the exact order in which these different sets of insertions occurred could not be determined.
The meta-cleavage pathway genes apparently have been assembled from different sources. The tdnG gene was separated from the other meta-cleavage pathway genes by a partial transposase gene and one of the two repeated sequences. Furthermore, the % G+C content of tdnG was significantly higher than the other meta-cleavage pathway genes. Novel pathways seem to form by an assembly process that involves horizontal gene transfer and recruitment of different parts of the novel pathway from different hosts into one single host (Top and Springael 2003). Hence, sequencing pCT14 may have revealed an intermediate stage in the evolution of a new assemblage of meta-cleavage pathway genes.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Bitton G (1994) Wastewater microbiology. Wiley-Liss, New York
Bramucci MG, Nagarajan V (2000) Industrial wastewater bioreactors: sources of novel microorganisms for biotechnology. Trends Biotechnol 18:501–505
Bramucci MG, McCutchen CM, Singh M, Thomas SM, Larsen BS, Buckholz J, Nagarajan V (2002a) Pure bacterial isolates that convert p-xylene to terephthalic acid. Appl Microbiol Biotechnol 58:255–259
Bramucci MG, Singh M, Nagarajan V (2002b) Biotransformation of p-xylene and 2,6-dimethylnaphthalene by xylene monooxygenase cloned from a Sphingomonas isolate. Appl Microbiol Biotechnol 59:679–684
Chattoraj DK (2000) Control of plasmid DNA replication by iterons: no longer paradoxical. Mol Microbiol 37:467–476
Cheng Q, Thomas SM, Kostichka K, Valentine JR, Nagarajan V (2000) Genetic analysis of a gene cluster for cyclohexanol oxidation in Acinetobacter sp. strain SE19 by in vitro transposition. J Bacteriol 182:4744–4751
Dogra C, Raina V, Pal R, Suar M, Lal S, Gartemann K, Holliger C, van der Meer JR, Lal R (2004) Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. J Bacteriol 186:2225–2235
Eckhardt T (1978) A rapid method for the identification of plasmid desoxyribonucleic acid in bacteria. Plasmid 1:584–588
Gardner MN, Deane SM, Rawlings DE (2001) Isolation of a new broad-host-range IncQ-like plasmid, pTC-F14, from the acidophilic bacterium Acidithiobacillus caldus and analysis of the plasmid replicon. J Bacteriol 183:3303–3309
Goebel M, Kranz OH, Kaschabek SR, Schmidt E, Pieper DE, Reineke W (2004) Microorganisms degrading chlorobenzene via a meta-cleavage pathway harbor highly similar chlorocatechol 2,3-dioxygenase-encoding gene clusters. Arch Microbiol 182:147–156
Greated A, Lambertsen L, Williams PA, Thomas CM (2002) Complete sequence of the IncP-9 TOL plasmid pWW0 from Pseudomonas putida. Environ Microbiol 4:856–871
Hofer B, Backhaus S, Timmis KN (1994) The biphenyl/polychlorinated biphenyl-degradation locus (bph) of Pseudomonas sp. LB400 encodes four additional metabolic enzymes. Gene 144:9–16
Igloi GL, Brandsch R (2003) Sequence of the 165-kilobase catabolic plasmid pAO1 from Arthrobacter nicotinovorans and identification of a pAO1-dependent nicotine uptake system. J Bacteriol 185:1976–1986
Kholodii GY, Yurieva OV, Lomovskaya OL, Gorlenko Z, Mindlin SZ, Nikiforov VG (1993) Tn5053, a mercury resistance transposon with integron's ends. J Mol Biol 230:1103–1107
Kholodii GY, Mindlin SZ, Bass IA, Yurieva OV, Minakhina SV, Nikiforov VG (1995) Four genes, two ends, and a res region are involved in transposition of Tn5053: a paradigm for a novel family of transposons carrying either a mer operon or an integron. Mol Microbiol 17:1189–1200
Kostichka K, Thomas SM, Gibson KJ, Nagarajan V, Cheng Q (2001) Cloning and characterization of a gene cluster for cyclododecanone oxidation in Rhodococcus ruber SC1. J Bacteriol 183:6478–6486
Lauf U, Muller C, Herrmann H (1998) The transposable elements resident on the plasmids of Pseudomonas putida strain H, Tn5501 and Tn5502, are cryptic transposons of the Tn3 family. Mol Gen Genet 259:674–678
Lawrence JG, Roth JR (1996) Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics 143:1843–1860
Leschziner AE, Boocock MR, Grindley ND (1995) The tyrosine-6 hydroxyl of gamma delta resolvase is not required for the DNA cleavage and rejoining reactions. Mol Microbiol 15:865–870
Li W, Shi J, Wang X, Han Y, Tong W, Ma L, Liu B, Cai B (2004) Complete nucleotide sequence and organization of the naphthalene catabolic plasmid pND6-1 from Pseudomonas sp. strain ND6. Gene 336:231–240
Maeda K, Nojiri H, Shintani M, Yoshida T, Habe H, Omori T (2003) Complete nucleotide sequence of carbazole/dioxin-degrading plasmid pCAR1 in Pseudomonas resinovorans strain CA10 indicates its mosaicity and the presence of large catabolic transposon Tn4676. J Mol Biol 326:21–33
Mars AE, Kingma J, Kaschabek SR, Reineke W, Janssen DB (1999) Conversion of 3-chlorocatechol by various catechol 2,3-dioxygenases and sequence analysis of the chlorocatechol dioxygenase region of Pseudomonas putida GJ31. J Bacteriol 181:1309–1318
Martinez B, Tomkins J, Wackett LP, Wing R, Sadowsky MJ (2001) Complete nucleotide sequence and organization of the atrazine catabolic plasmid pADP-1 from Pseudomonas sp. strain ADP. J Bacteriol 183:5684–5697
Minakhina S, Kholodii G, Mindlin S, Yurieva O, Nikiforov V (1999) Tn5053 family transposons are res site hunters sensing plasmidal res sites occupied by cognate resolvases. Mol Microbiol 33:1059–1068
Nojiri H, Shintani M, Omori T (2004) Divergence of mobile genetic elements involved in the distribution of xenobiotic–catabolic capacity. Appl Microbiol Biotechnol 64:154–174
Ochman H, Lawrence JG (1996) Phylogenetics and the amelioration of bacterial genomes. In: Neidhardt FC (ed) Escherichia coli and Salmonella: cellular and molecular biology, 2nd edn. ASM Press, Washington, DC, pp 2627–2637
Page DT, Whelan KF, Colleran E (2001) Characterization of two autoreplicative regions of the IncHI2 plasmid R478: RepHI2A and RepHI1A(R478). Microbiology 147:1591–1598
Paterson ES, Iyer VN (1997) Localization of the nic site of IncN conjugative plasmid pCU1 through formation of a hybrid oriT. J Bacteriol 179:5768–5776
Paterson ES, More MI, Pillay G, Cellini C, Woodgate R, Walker GC, Iyer VN, Winans SC (1999) Genetic analysis of the mobilization and leading regions of the IncN plasmids pKM101 and pCU1. J Bacteriol 181:2572–2583
Pullinger GD, Lax AJ (1992) Salmonella dublin virulence plasmid locus that affects bacterial growth under nutrient-limited conditions. Mol Microbiol 6:1631–1643
Radnedge L, Davis MA, Youngren B, Austin SJ (1997) Plasmid maintenance functions of the large virulence plasmid of Shigella flexneri. J Bacteriol 179:3670–3675
Romine MF, Stillwell LC, Wong KK, Thurston SJ, Sisk EC, Sensen C, Gaasterland T, Fredrickson JK, Saffer JD (1999) Complete sequence of a 184-kilobase catabolic plasmid from Sphingomonas aromaticivorans F199. J Bacteriol 181:1585–1602
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Sentchilo VS, Perebituk AN, Zehnder AJB, van der Meer JR, (2000) Molecular diversity of plasmids bearing genes that encode toluene and xylene metabolism in Pseudomonas strains isolated from different contaminated sites in Belarus. Appl Environ Microbiol 66:2842–2852
Thomas CM (2000) Paradigms of plasmid organization. Mol Microbiol 37:485–491
Top EM, Springael D (2003) The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr Opin Biotechnol 14:262–269
Vedler E, Vahter M, Heinaru A (2004) The Completely sequenced plasmid pEST4011 contains a novel IncP1 backbone and a catabolic transposon harboring tfd genes for 2,4-dichlorophenoxyacetic acid degradation. J Bacteriol 186:7161–7174
Venkova-Canova T, Soberon NE, Ramirez-Romero MA, Cevallos MA (2004) Two discrete elements are required for the replication of a repABC plasmid: an antisense RNA and a stem-loop structure. Mol Microbiol 54:1431–1444
Williams PA, Worsey MJ (1976) Ubiquity of plasmids in coding for toluene and xylene metabolism in soil bacteria: evidence for the existence of new TOL plasmids. J Bacteriol 125:818–828
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bramucci, M., Chen, M. & Nagarajan, V. Genetic organization of a plasmid from an industrial wastewater bioreactor. Appl Microbiol Biotechnol 71, 67–74 (2006). https://doi.org/10.1007/s00253-005-0119-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0119-2