
Abstract
We found a novel cyclodextrin glucanotransferase (CGTase) from alkalophilic Bacillus sp. G-825-6. The enzyme was expressed in the culture broth by recombinant Bacillus subtilis KN2 and was purified and characterized. The enzyme named CGTase825-6 showed 95% amino acid sequence identity with a known enzyme β-/γ-CGTase from Bacillus firmus/lentus 290-3. However, the product specificity of CGTase825-6 differed from that of β-/γ-CGTase. CGTase825-6 produced γ-cyclodextrin (CD) as the main product, but degradation of γ-CD was observed with prolonged reaction. The product specificity of the enzyme was positioned between γ-CGTase produced by Bacillus clarkii 7364 and B. firmus/lentus 290-3 β-/γ-CGTase. It showed that the difference of product specificity was dependent on only 28 amino acid residues in 671 residues in CGTase825-6. We compared the amino acid sequence of CGTase825-6 and those of other CGTases and constructed a protein structure model of CGTase825-6. The comparison suggested that the diminished loop (Val138-Asp142) should provide subsite -8 for γ-CD production and that Asp142 might have an important role in product specificity. CGTase825-6 should be a useful tool to produce γ-CD and to study the differences of producing mechanisms between γ-CD and β-CD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cyclodextrin glucanotransferase (CGTase; EC 2.4.1.19) is an enzyme that catalyzes the cleavage of α-1, 4-glycosidic bonds in starch and glycogen and intramolecular rearrangement (Qi and Zimmermann 2005). The resultant cyclic α-1, 4-linked oligosaccharides of six, seven, and eight glucose residues are named α-, β-, and γ-cyclodextrin (CD), respectively. CDs can form inclusion complexes with a wide variety of hydrophobic guest molecules, and the inclusion leads to changes in the chemical and physical properties of the guest molecules. Due to these properties, CDs are extensively utilized in the industry for various purposes (Biwer et al. 2002; van der Veen et al. 2000). CGTase is a member of the α-amylase family of glycosyl hydrolases (family 13), an important group of starch-converting enzymes. Whereas amylases generally hydrolyze glycosidic bonds in starch and glycogen, CGTase activity consists of four reactions (cyclization, coupling, disproportionation, and hydrolysis). The enzyme primarily catalyses transglycosylation reactions; hydrolysis activity is the minor reaction. The structure–function relationship in cyclization has been extensively studied. It has been reported that the enzyme from Bacillus circulans 251 had nine subsites from −2 to +7 (Strokopytov et al. 1996; Uitdehaag et al. 1999a,b, 2000; van der Veen et al. 2000). More than ten residues are considered to be concerned with substrate binding in the subsites. However, in spite of the energetic study on the CD synthesizing mechanism of CGTase, the mechanism by which the enzyme determines the size of synthesized CD is unclear, and the production of a single type of CD has been difficult (Biwer et al. 2002). In particular, there have been a few reports about γ-CGTase. As the cavity of the γ-CD ring is larger than those of α-CD and β-CD, the larger molecules that could not be trapped by α-CD and β-CD were entrapped in it.
Therefore, in this study, we cloned a novel CGTase from alkalophilic Bacillus sp. G-825-6, which is known as the alkaline subtilisin Sendai producing strain (Yamagata et al. 1995a). The enzyme produced γ-CD as the major product. The deduced amino acid sequence revealed that the enzyme had 95 and 70% of the identities of CGTase from Bacillus sp. 290-3 (Schmid 1991) and Bacillus clarkii 7364, respectively (Takada et al. 2003). CGTase 290-3 from Bacillus sp. 290-3 produced almost same the quantity of γ-CD as β-CD, and the enzyme was classified as a β-/γ-CGTase (Takada et al. 2003). In spite of sharing a high identity with β-/γ-CGTase 290-3, the major product of our enzyme was γ-CD; its optimum pH was higher than that of CGTase 290-3. The properties of the enzyme were different from B. clarkii 7364 γ-CGTase and β-/γ-CGTase 290-3. We discussed the property and the structure of the enzyme, subsite -7 in particular, and presumed those of subsite -8.
Materials and methods
Bacterial strains and plasmids
Alkalophilic Bacillus sp. G-825-6 that was used for the chromosomal DNA source has been described in our previous paper (Yamagata et al. 1995a,b). Escherichia coli DH5α [supE44, lacU169 (ϕ80 lacZΔM15) hsdR17, recA1, endA1, gyrA96, thi-1, relA1] was used for routine transformation. Bacillus subtilis KN2 (trpC2, phe-1, nprE18, aprEo3, ΔispA) was used for protein expression (Nakamura et al. 1990). Plasmids pUC118 and pUC119 were purchased from Takara Shuzo Co. Ltd. (Tokyo, Japan), and pBluescript II KS+ was purchased from Toyobo (Osaka, Japan).
DNA techniques
The following procedures were carried out by the standard methods described by Ausubel et al. (1987). Chromosomal DNA was prepared from alkalophilic Bacillus sp. as described by Saito and Miura (1963). DNA sequencing was carried out by using BigDye terminator ver. 3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA).
Screening of CGTase-producing recombinants
A degenerate pair of PCR primers designed from the protein sequence data of α-amylases from various Bacilli (primer Bf, 5′-GAYGGHTTYCGTVTTGATRCBG-3′; Cr, 5′-RTCATGRTTHTCNACRAAKG-3′) was used for partial starch-degrading enzyme gene fragment amplification using genomic DNA as a template. Primers inverse F (5′-GATAAACCGCAAGATCAAGTTACC-3′) and inverse R (5′-TCTTTTGCCACCCTACTGGC-3′) were used for inverse PCR to clone the full length of the starch-degrading enzyme gene from self-ligation libraries constructed with XbaI or EcoRI digests of genomic DNA. Finally, a 3.9-kbp fragment of genomic DNA digested with SpeI and SalI was inserted into pBluescript II KS+. The constructed partial genome library was introduced into E. coli DH5α. The transformants were plated on an LB agar plate containing 0.2% soluble starch and 50 μg/ml ampicillin–sodium salt, and the starch-degrading activity was detected with starch–iodine reaction by introducing iodine in the plate.
Construction of CGTase expression system with B. subtilis
The obtained genomic DNA fragment coding CGTase was digested with EcoRV and SalI, and the truncated fragment was inserted into pUC118 digested with SmaI and SalI. The resultant plasmid was designated as pUC118-CG. Plasmid pUB110 was digested with EcoRI and ligated with EcoRI fragment of pUC119 (pUBC119). The BamHI site, XbaI site, and one of the EcoRI sites in the resultant plasmid were deleted by the method proposed by Carter et al. (1985) by using 5′-TTGAGCAACTCGACCCAGGAGAACAAA-3′ and 5′-AGATTGATTTTTTGGAAAAAAATTTAG-3′, respectively. The plasmid was named pUBC119BX. Plasmid pUC118 was digested with EcoRI and ScaI, and a 1.4-kbp fragment was inserted into pUBC119BX partially digested with the same restriction endonucleases. The resultant plasmid was named pUBC1198BX. Plasmid pUC118-CG was digested with SacI and ligated with the SacI fragment of pUBC1198BX, and the resultant plasmid was designated as pCG-Bac. B. subtilis KN2 was transformed with the pCG-Bac using modified protoplast transformation. The protoplast transformation was performed with the method proposed by Chang and Cohen (1979).
Purification of CGTase from recombinant B. subtilis
B. subtilis KN2 carrying the expression plasmid was aerobically cultured in 100 ml of 2× LB containing 50 μg/ml kanamycin sulfate in a 500-ml Erlenmeyer flask with baffles at 37°C for 24 h. The culture broth was obtained by centrifuging at 12,000×g at 4°C for 20 min, and cornstarch was added to the supernatant up to 5% (w/w) and agitated overnight. The solution was filtered with a No. 2 filter paper (Advantec, Tokyo, Japan), and the residuum was washed with 10 mM citrate buffer (pH 5.5) containing 1 M NaCl and 2 mM CaCl2. The enzyme was eluted from the cornstarch solution with 10 mM citrate buffer containing 3 M NaCl, 0.5 M maltose, and 2 mM CaCl2. The eluted fraction was dialyzed against 10 mM citrate buffer (pH 5.5) containing 2 mM CaCl2 and applied to DEAE-Toyopearl 650 M (ϕ 2×8 cm) (Tosoh, Tokyo, Japan) equilibrated with the same buffer. The column was washed with the same buffer, and the enzyme was eluted with 0–0.1 M NaCl linear gradient. The enzyme active fraction was collected and added up to a 60% saturated concentration of ammonium sulfate. The mixture was applied to Octyl-cellulofine TypeS (ϕ 3.1×8 cm) and equilibrated with 10 mM citrate buffer (pH 5.5) containing 2 mM CaCl2 and 60% saturated concentration of ammonium sulfate. Elution was performed with 60–0% of saturated ammonium sulfate linear gradient. The active fraction was collected and pooled. In all purification steps, γ-CD synthesizing activity was measured.
Enzymatic activity assays
γ-CD synthesizing activity was estimated by the bromocresol green (BCG) method of Kato and Horikoshi (1984) with the following modification. One hundred microliters of 1.0% soluble starch in 100 mM acetate–citrate–borate buffer (pH 9.0) was preincubated at 37°C, and the adequate amount of the enzyme solution was added to the mixture. Following incubation for 40 min, 0.5 M citrate buffer (pH 4.0) was added to the reaction mix to stop the enzymatic reaction. Ten microliters of 5 mM BCG in 20% (v/v) ethanol was added to the mixture. One unit of γ-CD synthesizing activity was defined as the amount of the enzyme that yields the color equivalent to 1 μmol of γ-CD per min at pH 9.0 and 37°C.
Analysis of produced CDs
Ninety microliters of 0.1 M acetate–citrate–borate buffer, at each pH (4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, and 12.0), containing 1.0 and 10.0% of soluble starch was preincubated at 50°C; 10 μl of the enzyme solution (1.0 mU) was added and incubated at 50°C for 1 h. After incubation, the mixture was treated at 100°C for 5 min, and the heat-treated mixture was then left on ice. One hundred microliters of 0.5 M citrate buffer (pH 5.0) containing 160 U/ml glucoamylase was added to the mixture. The mixture was incubated at 45°C for 2 h, and the enzyme was then inactivated by heat treatment at 100°C for 5 min. The treated mixture was centrifuged at 18,500×g at 4°C for 5 min. The supernatant was lyophilized. The sample was dissolved in 50 μl of MilliQ water, and 20 μl of the solution was applied to a TSKgel Amide-80 column (4.6×250 mm, Tosoh) equipped with Shimadzu LC-6A high-pressure liquid chromatography (HPLC) system (Shimadzu Co., Kyoto, Japan). The elution was performed with 60% of acetonitrile, and the flow rate was observed to be 0.6 ml/min. The eluted CDs from the column were detected with a refractive index detector R401 (Japan Waters Co., Tokyo, Japan).
Electrophoresis of proteins
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was performed using the method of Laemmli (1970) on a 12.0% polyacrylamide slab gel, and the protein bands were stained with Coomassie brilliant blue R-250. The molecular mass standards used were from New England Biolabs (Beverly, MA, USA).
N-terminal amino acid sequence
The purified enzyme was applied to SDS-PAGE, and the protein band was transferred to polyvinylidene fluoride (PVDF) membrane using a semi-dry electroblotter (Trans-Blot SD, Bio-Rad Laboratories, Hercules, CA, USA). The Coomassie brilliant blue R-250 stained band was cut from the membrane, and its N-terminal sequence was analyzed on Applied Biosystems 473A protein sequencer with 610A data analysis system (Matsudaira 1987).
Protein concentration
Protein concentration was determined with BCA Protein Assay Kit (Pierce Biotechnology Inc., Rockford, IL, USA) (Smith et al. 1985). Bovine serum albumin included in the assay kit was used as a standard.
Protein structure modeling
Comparative protein structure modeling was performed with SWISS-MODEL (Schwede et al. 2003), and the protein structures of B. circulans 251 E257Q/D229N-CGTase (Uitdehaag et al. 1999a,b, 2002) and B. circulans No. 8 Δ(145–151)D-CGTase (Parsiegla et al. 1998) were used as homology models.
Nucleotide accession number
The nucleotide sequence of cgtS from alkalophilic Bacillus sp. G-825-6 has been assigned the DDBJ accession number AB201304.
Results
Cloning of the starch degradation enzyme gene
We obtained an 8,387-bp DNA fragment from the genomic DNA of Bacillus sp. G-825-6 digested with SpeI and SalI. There were two open reading frames (ORFs) in the fragment. The two ORFs consisted of 981 and 2,100 bp and encoded 325 and 699 amino acids, respectively. According to the results of the sequencing, the former should be the gene encoding a transposase, and the latter encoded CGTase. Transposase showed 96% identity with the transposase (Takami et al. 1999) from Bacillus halodulans (BAB07704). We designated the protein as transposes825-6, and the gene was named tpsS.
In the gene encoding CGTase, a potential Shine–Dalgarno (SD) sequence AAGGATGG (Gold et al. 1981) was found 8 bp upstream from a probable translation start codon (ATG). A promoter motif sequence (−10 region, TATTTT and TATATT; −35 region, TTGGCA and TTGCCA) was found at 55, 173, 95, and 226 bp of the SD sequence upstream, respectively (Moran et al. 1982). The amino acid sequence is also shown in Fig. 1. The first 28 amino acid residues were calculated as signal peptide region by the SignalP server (http://www.cbs.dtu.dk/services/SignalP). The molecular mass of the predicted mature region of 671 amino acid residues was estimated as 75,256 Da. The deduced mature sequence showed 70 and 95% identities with B. clarkii γ-CGTase (Takada et al. 2003) and B. firmus/lentus β-/γ-CGTase (Schmid 1991), respectively. The enzyme also consisted of amylolytic enzyme conserved regions (regions I, II, III, and IV), and regions II and III each contained a catalytic residue of Asp. We designated the enzyme as CGTase825-6, and the gene was named cgtS (Fig. 2).

Nucleotide sequence of cgtS and deduced amino acid sequence of CGT825-6 form Bacillus sp. G-825-6. The putative ribosome-binding site (SD sequence) is double underlined. The two putative promoters (−10 and −35 regions) are boxed. The underlined amino acid sequence was the predicted signal peptide; the succeeding amino acid sequence coincided with the N-terminal amino acid sequence of purified CGTase825-6. Full nucleotide sequence data reported in this paper will appear in the DDBJ/EMBL/GenBank nucleotide sequence with accession number AB201304

Amino acid sequence of the mature CGT825-6 and other bacterial CGTases. The four conserved regions in the α-amylase family are enclosed in open boxes. The amino acid sequence conserved in only the γ-CD-producing enzyme in comparison with 22 CGTases from B. circulans 251 (accession number; A58800), Bacillus sp. B1018 (P17692), Bacillus sp. 17-1 (P30921), B. circulans A-11 (AAG31622), Bacillus sp. 1011 (P05618), Bacillus sp. 38-2 (ALBSG3), Bacillus sp. 1-5 (AAR32682), B. circulans No. 8 (CAA48401), Bacillus sp. 6.3.3 (P31747), Bacillus licheniformis (P14014), P. macerans (AAC04359), P. macerans (P04830), Bacillus ohbensis (BAA14289), Bacillus sp. 1-1 (P31746), Thermosulfurigenesi EM1 (1A47), Geobacillus stearothermophilus (1CYG), Bacillus agaradhaerens LS-3C (AAP31242), B. agaradhaerens (CAD23265), B. agaradhaerens DMS 9948 (CAD38091), B. firmus/lentus 290-3 (CAA01436), and B. clarkii 7364 (BAB91217) are enclosed in dark boxes. The amino acid residues of CGTase 825-6 that differ from those of B. firums/lentus 290-3 CGTase are enclosed in the open circle
Purification of CGTase 825-6
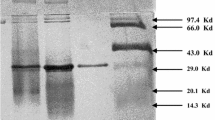
Purification steps of the enzyme are summarized in Table 1. We obtained the enzyme that had γ-CD-producing ability from the culture broth of recombinant B. subtilis carrying pCG-Bac. The purified enzyme showed 5.57 U/mg of specific activity for γ-CD production. SDS-PAGE of the purified enzyme is shown in Fig. 3. A single band was observed, and the molecular mass of the enzyme was estimated as 78.2 kDa. The N-terminal amino acid sequence of the enzyme was determined as 1Asn-Glu-Asn-Leu-5Asp-Asn-Val-Asn-Tyr-10Ala-Glu-Glu-Ile-Ile-15Tyr-. It coincided with the deduced amino acid sequence of the mature enzyme.

SDS-PAGE purified CGTase expressed by B. subtilis KN2. Lane 1 Molecular mass standards (New England Biolabs), lane 2 purified CGTase825-6
Properties of γ-CGTase
The optimum pH and product specificity were estimated with 1.0 mU of the enzyme and 1 and 10% of soluble starch as a substrate at each pH (pH 4.0–12.0) for 1 h (Fig. 4). α-CD production was not observed at any pH examined. The CD-producing activity was not observed at pH 4.0 and 12.0 with 1% soluble starch as a substrate, but the enzyme showed γ-CD synthesis with 10% substrate even at pH 4.0. The enzyme produced γ-CD principally at any pH, and the ratios of γ-/β-CD were always more than 1.7 and 4.7 with 1 and 10% substrate, respectively. The increment of substrate concentration caused the enlargement of γ-CD production and the decrease in β-CD production. The optimum pH for γ-CD production of the enzyme with 1% substrate was pH 9.0, but the optimum pH with 10% soluble starch was pH 8.0–10.0. On the other hand, β-CD-producing activity showed increment up to pH 8.0, but it was decreased at higher pH, i.e., more than 9.0, with both substrate concentrations. The optimum temperature was 50–55°C (data not shown). The pH stability of the enzyme was determined by assaying the residual activity after incubation at each pH (4.0–12.0) at 37°C for 30 min, and the stable pH was observed as pH 7.0–12.0 (data not shown). Thermostability of the enzyme was also determined by measuring the residual activity after incubating the enzyme at pH 5.5 at each temperature (4–60°C) for 1 h; the enzyme showed more than 90% of the original activity up to 50°C (data not shown).

Effect of pH and substrate concentration on CD production of CGTase. A reaction mixture containing 1.0, 5.0, or 10.0% soluble starch and purified γ-CGTase825-6 (780 μU) in 0.1 M acetate–borate–phosphate buffer (pH 4.0, 5.0, 6.0, 7.0, 8.0, 9.0, 10.0, 11.0, and 12.0) was incubated at 37°C. CDs formed in the mixture were determined by HPLC, as described in Materials and methods. Open boxes, dark boxes, and closed boxes indicate α-CD, β-CD, and γ-CD, respectively
Time course of CD production
The time course of CD-producing activity was measured with 2.0 mU of the enzyme at pH 10.0 and 11.0 at 50°C for 72 h (Fig. 5). At pH 10.0, CD production rate increased linearly for the first 4 h, but after that, the rate of the increment was diminished, and γ-CD was decreased after 24 h linearly. At pH 11.0, CD production rate was lesser than that at pH 10.0, and it started to plateau after 24 h. γ-CD began to decrease after 36 h. On the other hand, β-CD was increased continuously for 72 h at both pH. In either case, α-CD was not observed.

Time course of formation of CDs from soluble starch by γ-CGTase825-6. A reaction mixture containing 10% soluble starch and purified γ-CGTase825-6 (78 mU/g of dry starch) in 0.1 M acetate–borate–phosphate buffer (pH 10.0 or 11.0) was incubated at 37°C. CDs formed in the mixture were determined by HPLC, as described in Materials and methods
Discussion
We found a new CGTase from alkalophilic Bacillus sp. G-825-6 and named it CGTase825-6. The strain has been known to produce alkaline subtilisin Sendai (Yamagata et al. 1995a). We cloned and sequenced the gene encoding the CGTase, and the gene was designated as cgtS. CGTase825-6 was purified from recombinant B. subtilis KN2 carrying expression plasmid pCG-Bac, and the enzyme was confirmed to be γ-CGTase. The molecular mass of the purified enzyme determined by SDS-PAGE did not agree with the calculated molecular mass from the deduced amino acid sequence in spite of several trials of SDS-PAGE. Some alkaline proteases from alkalophilic Bacillus, including Bacillus sp. G-825-6, indicate larger molecular masses in SDS-PAGE than those from the calculated data (Yamagata et al. 1995a,b). There might be some characteristic structure in those alkaline enzymes.
As shown in Fig. 4, CGTase825-6 was the enzyme that hydrolyzed starch and produced γ-CD as a main product; however, the enzyme could not produce α-CD. Optimum pH of the CD production was in alkaline region. However, the optimum pH of γ-CD-producing activity and that of β-CD were different from each other with 10% soluble starch as a substrate. The optimum pH of β-CD-producing activity was at pH 8.0, but γ-CD production showed optimum pH at pH 8.0-10.0. β-CD production was restricted under higher alkaline conditions with 10% substrate. The differences may have been caused by the subtle structural change due to alkaline pH. As the cyclizing reaction was observed at alkaline pH (because γ-CD was produced), substrate-binding modes of the enzyme should be different between pH 8.0 and pH 9.0–10.0.
Concentration of the substrate showed drastic change of the producing rate of β- and γ-CD. β-CD-producing activity was decreased by the increment of the substrate concentration, but high-concentration substrate induced acceleration of γ-CD production of the CGTase825-6. The results indicated that the concentration of the substrate also affected the substrate-binding mode of the enzyme. In the time-laps experiment with 10% substrate at pH 10.0 and 11.0, it was observed that the product specificity of the CGT825-6 placed a disproportionate emphasis on γ-CD production, but γ-CD was decreased in reaction mixture for the longer reaction (Fig. 5). The enzymatic reaction reached a plateau at a certain level of γ-CD accumulation at both pH 10.0 and 11.0, and then the γ-CD was decreased linearly. It was presumed that the γ-CD might be digested by CGTase825-6 itself. As it was reported that B. clarkii CGTase could not digest γ-CD and γ-CD would inhibit the CD-producing reaction of the enzyme (Takada et al. 2003), we examined the γ- and β-CD degrading activity of CGTase825-6 and found out that the enzyme could degrade both CDs (data not shown). The results suggest that the decrease in γ-CD was the result of the degradation of the CD by CGTase825-6. However, β-CD did not decrease in the reaction mixture. The results also suggested that γ-CD was a more preferable substrate than β-CD for CGTase825-6. As hydrolysis reaction mechanism of both CDs should not be different, γ-CD could bind to the substrate-binding site of the enzyme more easily than β-CD could. The substrate-binding site of the enzyme should be suitable for γ-CD. It was presumed that there must be subsites for eight glucose residues, and eight glucose residues should be necessary for the stable substrate–enzyme complex rather than seven residues.
Most of the innumerous reports on CGTase are about α- or β-CGTase (Qi and Zimmermann 2005; Biwer et al. 2002; van der Veen et al. 2000), whereas there are few reports on γ-CGTase. CGTase from B. clarkii 7364 is classified as γ-CGTase (Takada et al. 2003), and the enzyme from B. firmus/lentus 290-3 is classified as β-/γ-CGTase (Takada et al. 2003). CGTase 825-6 was distinguishable from these enzymes because its product specificity was different. CGTase825-6 could not produce α-CD, but B. clarkii CGTase showed α-CD production. γ-CD was the main product of CGTase825-6, but B. firmus/lentus CGTase produced both β- and γ-CD equally even in the initial reaction. The optimum pH for CGTase825-6 was more than 8.0 with any substrate concentration, but B. firmus/lentus CGTase had an optimum pH in the range of 6.0–8.0. To clarify the mechanism of product specificity, it is important to compare the amino acid sequences of the CGTases. On the basis of the results of many studies on the x-ray crystallographic structures of CGTases with their substrates, inhibitors, or products (Harata et al. 1996; Lawson et al. 1994; Strokopytov et al. 1996; Uitdehaag et al. 1999a,b, 2000), it has been proposed that the active center of CGTase has a tandem subsite architecture in the substrate-binding groove and that it contains at least nine sugar-binding subsites designated, from the non-reducing end to the reducing end, as −7 through +2 (Qi and Zimmermann 2005). Several amino acid residues that are involved in the binding of maltononaose (Strokopytov et al. 1996) or γ-CD (Uitdehaag et al. 1999a,b) with β-CGTase from B. circulans 251 have been revealed. We compared the amino acid sequence of CGTase825-6 with those of 21 other CGTases (accession numbers A58800, P17692, P30921, AAG31622, P05618, ALBSG3, AAR32682, CAA48401, P31747, P14014, AAC04359, P04830, BAA14289, P31746, 1A47, 1CYG, AAP31242, CAD23265, CAD38091, CAA01436, and BAB91217). The alignment of α-CGTase from Paenibacillus macerans (Takano et al. 1986), β-CGTase from B. circulans 251 (Lawson et al. 1994), β-/γ-CGTase from B. firmus/lentus 290-3, γ-CGTase from B. clarkii 7364, and CGTase825-6 from Bacillus sp. G-825-6 are shown in Fig. 2. It shows that there are four conserved regions in all CGTases, which are conserved in α-amylase family, but 26 amino acid residues were conserved only in the γ-CD-producing enzyme originally. Furthermore, there were a few amino acid residues that are concerned with substrate-binding sites in these originally conserved 26 amino acid residues. An amino acid residue positioned at 47 (B. circulans 251 CGTase numbering) was reported as subsite −3 forming residue, and a basic amino acid residue, such as Lys, Arg, or His, occupies the position in α- and γ-CGTases. However, the residue was commonly observed in three γ-CD-producing enzymes. In subsite −3, the deletion of three residues (LHPGGFA-S-) was also observed in positions 85–96 (VIKYSGVNNTS and IINYSGVNNTA in α- and β-CGTase, respectively). In subsite -7, there was a six-residue deletion (VDIEN/D-) in the region 144–154 (ADRDNPGFAEN and ASSDQPSFAEN in α- and β-CGTase, respectively). We focused attention on the deletion in the region 144–154 in the primary structure of β-CGTase. As described above, the region 144–154 is concerned with forming subsite −7 in β-CGTase. The aspect of maltononaose in subsite −7 of B. circulans 251 β-CGTase (Strokopytov et al. 1996) (Fig. 6a) superimposed B. circulans No. 8 Δ(145–151)D-CGTase (Parsiegla et al. 1998) (Fig. 6b) and superimposed the constructed model of CGTase825 (Fig. 6c), as shown in Fig. 6. It was reported that β-CGTase from B. circulans 251 had a big loop formed in subsite −7, and Ser145 and Asp147 on the loop interacted with the glucose residue bound to subsite −7. B. circulans No. 8 Δ(145–151)D-CGTase is a mutant enzyme that deletes amino acid residues of 145–151 and inserts a single Asp. Because of the mutation, the enzyme lacks the loop structure of 144–154, and the production rate of β-/γ-CD of the enzyme is converted from 64/20 to 54/40. It is considered that the Δ(145–151)D mutant enzyme obtains more space for binding the eighth glucose residue due to the loss of the loop. Furthermore, the inserted Asp151, corresponding to Ser151 of β-CGTase, interacts with the seventh and eighth glucose residue in the subsite, respectively. As a result, the substrate α-1, 4-glucan should be able to bind not only at subsite −7 but also at presumed subsite −8, and the γ-CD-producing activity should be increased. As in the case of Δ(145–151)D-CGTase, CGT825-6 had enough space for the subsite −8 by deletion in the loop. However, unlike Δ(145–151)D-CGTase, the diminished loop of CGTase825-6 formed the wall of subsite −8. The wall might prevent the binding of the ninth glucose residue or a longer substrate and stabilize the interaction between a glucose residue and subsite −8. Furthermore, we presumed that amino acid residues on the loop might be concerned with the stabilization of the interaction, and Asp142 (CGTase825 numbering) on the small loop was one of the candidates. The residue was conserved in CGTase825-6 and B. clarkii γ-CGTase, but the corresponding residue in B. firmus/lentus β-/γ-CGTase was Asn. The amino acid sequence of CGTase825-6 showed 95% identity with that of B. firmus/lentus 290-3 CGTase; therefore, only 29 amino acid residues out of 671 were different between the two enzymes. Nevertheless, as described above, their product specificity was markedly different from each other. CGTase825-6 produced only γ-CD, whereas B. firmus/lentus 290-3 CGTase produced both β- and γ-CD under a reaction condition. As it was believed that B. firmus/lentus 290-3 CGTase was a natural mutant of CGTase825-6, we compared the amino acid sequences of the two CGTases. All other different residues were found in the regions or sites concerned with active center or substrate-binding site previously reported, except for Asp142. Because of it, the residue might be important for product specificity. The Δ(145–151)D-CGTase might not have enough length of loop to form subsite −8. As a result, the production rate of γ-CD did not increase as compared with γ-CGTase. Furthermore, the mutant might not have a corresponding residue with Asp142 of CGTase825-6. A short loop and an adequate residue interacting with the glucose residue in this region should be necessary to form subsite −8 and produce γ-CD.

Stereo pictures indicating the maltononoase (white) and subsite −7 in the CGTase (dark gray) of B. circulans 251 β-CGTase (Strokopytov et al. 1996) (a), the model of CGTase825-6 (this study) (b), and B. circulans No. 8 Δ(145–151)D-CGTase (Parsiegla et al. 1998) (c). Each maltononaose in (b) and (c) is superimposed using the SWISS-MODEL software (Schwede et al. 2003). The glycosyl residues of maltononaose are numbered −5, −6 and −7 as their corresponding subsite numberings
As described above, we found a new CGTase that produced γ-CD as a main product. As the enzyme γ-CGTase825-6 showed high homology with β-/γ-CGTase from B. firmus/lentus in spite of a difference in product specificity, we believed that the enzyme should be a good tool for the application and the study of γ-CD production.
References
Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1987) Current protocols in molecular biology. Greene and Wiley, New York
Biwer A, Antranikian G, Heinzle E (2002) Enzymatic production of cyclodextrins. Appl Microbiol Biotechnol 59:609–617
Carter P, Bedouelle H, Winter G (1985) Improved oligonucleotide site-directed mutagenesis using M13 vectors. Nucleic Acids Res 13:4431–4443
Chang S, Cohen SN (1979) High frequency transformation of Bacillus subtilis protoplasts by plasmid DNA. Mol Gen Genet 168:111–115
Gold L, Pribnow D, Schneider T, Shinedling S, Singer BS, Stormo G (1981) Translation initiation in prokaryotes. Annu Rev Microbiol 35:365–403
Harata K, Haga K, Nakamura A, Aoyagi M, Yamane M (1996) X-ray structure of cyclodextrin glucano-transferase from alkalophilic Bacillus Sp. 1011. Comparison of two independent molecules at 1.8 Å resolution. Acta Crystallogr, D Biol Crystallogr 52:1136–1145
Kato K, Horikoshi K (1984) Colorimetric determination of gamma-cyclodextrin. Agric Biol Chem 56:1738–1740
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lawson CL, van Montfort R, Strokopytov B, Rozeboom HJ, Kalk KH, de Vries GE, Penninga D, Dijkhuizen L, Dijkstra BW (1994) Nucleotide sequence and X-ray structure of cyclodextrin glycosyltransferase from Bacillus circulans strain 251 in a maltose-dependent crystal form. J Mol Biol 236:590–600
Matsudaira P (1987) Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem 262:10035–10038
Moran C, Lang N, LeGrice S, Lee G, Stephans M, Sonenshein A, Pero J, Losick R (1982) Nucleotide sequences that signal the initiation of transcription and translation in Bacillus subtilis. Mol Gen Genet 186:339–346
Nakamura A, Koide Y, Kawamura F, Horinouchi S, Uozumi T, Beppu T (1990) Construction of a Bacillus subtilis strain deficient in three proteases. Agric Biol Chem 54:1307–1309
Parsiegla G, Schmidt AK, Schulz GE (1998) Substrate binding to a cyclodextrin glycosyltransferase and mutations increasing the gamma-cyclodextrin production. Eur J Biochem 255:710–717
Qi Q, Zimmermann W (2005) Cyclodextrin glucanotransferase: from gene to applications. Appl Microbiol Biotechnol 66:475–485
Saito H, Miura K (1963) Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim Biophys Acta 72:619–629
Schmid G (1991) Gammer-CGTase. US Patent 5,409,824
Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31:3381–3385
Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76–85
Strokopytov B, Knegtel RM, Penninga D, Rozeboom HJ, Kalk KH, Dijkhuizen L, Dijkstra BW (1996) Structure of cyclodextrin glycosyltransferase complexed with a maltononaose inhibitor at 2.6 angstrom resolution. Implications for product specificity. Biochemistry 35:4241–4249
Takada M, Nakagawa Y, Yamamoto M (2003) Biochemical and genetic analyses of a novel gamma-cyclodextrin glucanotransferase from an alkalophilic Bacillus clarkii 7364. J Biochem (Tokyo) 133:317–324
Takami H, Takaki Y, Nakasone K, Sakiyama T, Maeno G, Sasaki R, Hirama C, Fuji F, Masui N (1999) Genetic analysis of the chromosome of alkaliphilic Bacillus halodurans C-125. Extremophiles 3:227–233
Takano T, Fukuda M, Monma M, Kobayashi S, Kainuma K, Yamane K (1986) Molecular cloning, DNA nucleotide sequencing, and expression in Bacillus subtilis cells of the Bacillus macerans cyclodextrin glucanotransferase gene. J Bacteriol 166:1118–1122
Uitdehaag JC, Mosi R, Kalk KH, van der Veen BA, Dijkhuizen L, Withers SG, Dijkstra BW (1999a) X-ray structures along the reaction pathway of cyclodextrin glycosyltransferase elucidate catalysis in the alpha-amylase family. Nat Struct Biol 6:432–436
Uitdehaag JC, Kalk KH, van der Veen BA, Dijkhuizen L, Dijkstra BW (1999b) The cyclization mechanism of cyclodextrin glycosyltransferase (CGTase) as revealed by a gamma–cyclodextrin–CGTase complex at 1.8-A resolution. J Biol Chem 274:34868–34876
Uitdehaag JC, van Alebeek GJ, van Der Veen BA, Dijkhuizen L, Dijkstra BW (2000) Structures of maltohexaose and maltoheptaose bound at the donor sites of cyclodextrin glycosyltransferase give insight into the mechanisms of transglycosylation activity and cyclodextrin size specificity. Biochemistry 39:7772–7780
van der Veen BA, Uitdehaag JC, Dijkstra BW, Dijkhuizen L (2000) Engineering of cyclodextrin glycosyltransferase reaction and product specificity. Biochim Biophys Acta 1543:336–360
Yamagata Y, Isshiki K, Ichishima E (1995a) Subtilisin Sendai from alkalophilic Bacillus sp.: molecular and enzymatic properties of the enzyme and molecular cloning and characterization of the gene, aprS. Enzyme Microb Technol 17:653–663
Yamagata Y, Sato T, Hanzawa S, Ichishima E (1995b) The structure of subtilisin ALP I from alkalophilic Bacillus sp. NKS-21. Curr Microbiol 30:201–209
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hirano, K., Ishihara, T., Ogasawara, S. et al. Molecular cloning and characterization of a novel γ-CGTase from alkalophilic Bacillus sp.. Appl Microbiol Biotechnol 70, 193–201 (2006). https://doi.org/10.1007/s00253-005-0041-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-005-0041-7