
Abstract
The microbial degradation of polychlorinated biphenyls (PCBs) has been extensively studied in recent years. The genetic organization of biphenyl catabolic genes has been elucidated in various groups of microorganisms, their structures have been analyzed with respect to their evolutionary relationships, and new information on mobile elements has become available. Key enzymes, specifically biphenyl 2,3-dioxygenases, have been intensively characterized, structure/sequence relationships have been determined and enzymes optimized for PCB transformation. However, due to the complex metabolic network responsible for PCB degradation, optimizing degradation by single bacterial species is necessarily limited. As PCBs are usually not mineralized by biphenyl-degrading organisms, and cometabolism can result in the formation of toxic metabolites, the degradation of chlorobenzoates has received special attention. A broad set of bacterial strategies to degrade chlorobenzoates has recently been elucidated, including new pathways for the degradation of chlorocatechols as central intermediates of various chloroaromatic catabolic pathways. To optimize PCB degradation in the environment beyond these metabolic limitations, enhancing degradation in the rhizosphere has been suggested, in addition to the application of surfactants to overcome bioavailability barriers. However, further research is necessary to understand the complex interactions between soil/sediment, pollutant, surfactant and microorganisms in different environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Polychlorinated biphenyls (PCBs) are a class of compounds where the aromatic biphenyl skeleton carries between one and ten chlorine atoms and, theoretically, 209 different PCB congeners can thus be produced. Typical commercial PCB mixtures, marketed as Aroclor, Kaneclor, Clophen or Delor, usually contain 20–60 congeners, predominantly tri-hexachlorinated congeners. Due to their non-flammability, chemical stability, high boiling point and electrical insulating properties, PCBs have been used for hundreds of industrial and commercial purposes, such as in electrical applications, heat transfer, and hydraulic equipment; as plasticizers in paints, plastics and rubber products; in pigments, dyes and carbonless copy paper, and many other applications. More than 1.5 million tons of PCBs were manufactured worldwide between 1927 and the early 1980s (Faroon et al. 2003), a significant amount of which has been released to the environment. The first adverse health effects were recorded in the 1930s (Drinker et al. 1937). Since then, PCBs have been shown to cause cancer in animals (Mayes et al. 1998), and also to cause a number of serious effects on the immune, reproductive, nervous and endocrine systems, among others (ATSDR 2000; Aoki 2001; Faroon et al. 2001). Studies in humans provide supportive evidence for the potential carcinogenic and non-carcinogenic effects of PCBs (ATSDR 2000).
Lunt and Evans (1970) and Catelani et al. (1970) were the first to isolate microorganisms capable of growing on biphenyl as sole source of carbon and energy and since then several biphenyl-degrading organisms have been isolated. These organisms belong to both Gram-negative and Gram-positive genera and comprise various Pseudomonas, Burkholderia, Achromobacter, Comamonas, Ralstonia, Acinetobacter, Rhodococcus and Bacillus isolates.
The degradation of biphenyl by microorganisms isolated thus far is initiated by a biphenyl 2,3-dioxygenase. Biphenyl 2,3-dioxygenases belong to a large family of Rieske non-heme iron oxygenases (Gibson and Parales 2000). They comprise a terminal oxygenase composed of a large α- and a small β-subunit, a ferredoxin and a ferredoxin reductase. The ferredoxin and ferredoxin reductase act as an electron transport system to transfer electrons from NADH to the terminal oxygenase. The cis-2,3-dihydro-2,3-dihydroxybiphenyl formed by this reaction is dehydrogenated by a dehydrogenase to give 2,3-dihydroxybiphenyl, which is subject to extradiol cleavage. The 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate thereby formed undergoes hydrolysis, yielding benzoate and 2-hydroxypenta-2,4-dienoate as reaction products. This sequence of reactions forms the so-called biphenyl upper pathway (Fig. 1). Ahmed and Focht (1973a, b) were the first to describe that biphenyl-degrading organisms have the capacity to transform several PCB congeners. Since then, a major focus of research on biphenyl-degrading organisms was their capability to transform PCBs, which differs significantly between different isolates.

Pathway for biphenyl degradation and genetic organization of the bph gene clusters of Burkholderia sp. LB400 (Erickson and Mondello 1992; Hofer et al. 1994), Pseudomonas putida KF715 (Hayase et al. 1990; Nishi et al. 2000), Rhodococcus sp. strain M5 (Peloquin and Greer 1993; Labbe et al. 1997), Rhodococcus sp. strain RHA1 (Masai et al. 1995; Takeda et al. 2004) and Achromobacter georgiopolitanum KKS102 (Kimbara et al. 1989; Kikuchi et al. 1994; Ohtsubo et al. 2001)
The biphenyl “upper pathway”
Genetic organization
Genes encoding enzymes of the biphenyl upper pathway (termed bph) were first cloned from Pseudomonas pseudoalcaligenes KF707 (Furukawa and Miyazaki 1986) and later from Burkholderia sp. LB400 (Mondello 1989). In both strains, the genes encoding for biphenyl 2,3-dioxygenase (bphA1A2A3A4), cis-2,3-dihydro-2,3-dihydroxybiphenyl dehydrogenase (dihydrodiol dehydrogenase, bphB), 2,3-dihydroxybiphenyl 1,2-dioxygenase (bphC) and 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase (HOPDA hydrolase, bphD) are organized in an operon, with genes encoding a glutathione S-transferase and enzymes involved in the transformation of 2-hydroxypenta-2,4-dienoate released during hydrolysis of HOPDA to form benzoate (2-hydroxypenta-2,4-dienoate hydratase, BphH; acetaldehyde dehydrogenase, BphJ; 4-hydroxy-2-oxovalerate aldolase, BphI) being localized between bphC and bphD (Hofer et al. 1994) (Fig. 1). A more detailed analysis revealed that such an organization occurs also in Ralstonia eutropha H850 (Bedard et al. 1987) and various other isolates (Bartels et al. 1999). Pseudomonas putida KF715 (Hayase et al. 1990) contains a simple bphABCD gene cluster (Fig. 1) and sequence comparisons have shown that this gene cluster still contains 106 nucleotides of a 2-hydroxy-2-oxovalerate aldolase-encoding gene, suggesting that this strain used to have the entire bph gene cluster as in LB400 (Nishi et al. 2000). The catabolic genes in LB400, KF707 and KF715 are preceded by a so-called orf0, and its gene product (Watanabe et al. 2003), belonging to the GntR family, was shown to positively regulate both its own expression and that of bphKHIJ in the presence of biphenyl in KF707 (Watanabe et al. 2000). HOPDA was proposed as the inducer. However, whereas orf0 mutants of KF707 failed to grow on biphenyl, and accumulated high levels of HOPDA due to the failure to express bphD, orf0 mutants of LB400 retained the ability to grow on biphenyl, but produced decreased concentrations of bphA1A2 RNA (Beltrametti et al. 2001). Thus, orf0 seems to be involved in expression of bphA genes in LB400. Whether orf0 is also involved in expression of bphA in KF707 remains to be elucidated. Recently, a second regulator, BphR2, belonging to the LysR family, was shown to act as a positive regulator activating transcription of bphA1A2A3A4BC in KF707 (Watanabe et al. 2003).
A second type of bph gene cluster was observed in Achromobacter georgiopolitanum (formerly Pseudomonas sp.) strain KKS102 (Kimbara et al. 1989; Kikuchi et al. 1994) and Wautersia oxalatica A5 (Springael et al. 1993; Merlin et al. 1997; Mouz et al. 1999) (Fig. 1). In these gene clusters, the order of bph genes differs significantly from that found in LB400, with genes encoding 2-hydroxypenta-2,4-dieneoate hydratase, acetaldehyde dehydrogenase, and 4-hydroxy-2-oxovalerate aldolase (designated bphEGF) preceding genes encoding upper pathway enzymes. Furthermore, the gene encoding the reductase subunit of biphenyl dioxygenase (bphA4) is localized at the end of the gene cluster, separated from bphD by an orf1 of unknown function. Regulation of bph genes in strain KKS102 was shown to be dependent on the product of the bphS gene located upstream of bphE. Its deduced amino acid sequence showed homology with the GntR family of transcriptional repressors. Disruption of bphS resulted in constitutive expression of bph genes, suggesting that the bphS gene product negatively regulated the pE promoter located upstream of the bphE gene. Binding of BphS was abolished in the presence of HOPDA (Ohtsubo et al. 2001).
A third type of gene cluster was observed in biphenyl-degrading Rhodococcus strains M5 (Peloquin and Greer 1993), RHA1 (Masai et al. 1995) and TA421 (Arai et al. 1998) (Fig. 1). The catabolic gene clusters differ slightly in their arrangements, and whereas the gene encoding dihydrodiol dehydrogenase follows that encoding 2,3-dihydroxybiphenyl dioxygenase in RHA1 (bphA1A2A3A4CB), it precedes the 2,3-dihydroxybiphenyl dioxygenase-encoding gene in TA421 (bphA1A2A3A4BC) and M5 (termed bpdC1C2BADE). However, in all three strains, this catabolic gene cluster is followed by bphS and bphT genes (termed bpdS and bpdT in strain M5) (Labbe et al. 1997; Arai et al. 1998; Takeda et al. 2004) that are believed to encode two-component signal transduction systems. BphT has a typical response regulator sequence, whereas BpdS, the predicted sensor kinase, is an unusually large transmembrane protein that contains ATP-binding and leucine-rich repeat motifs and some conserved residues of protein kinases. Expression of bpdST, like that of the bpdC1C2BADE degradative operon, is inducible by biphenyl (Labbe et al. 1997). The bphS and bphT genes promote transcriptional induction by a variety of aromatic compounds in RHA1, including biphenyl, benzene, alkylbenzenes, and chlorinated benzenes (Takeda et al. 2004). However, 4-chlorobiphenyl was only a poor inducer.
Mobile genetic elements
The bphA1A2A3A4CBST gene cluster of Rhodococcus sp. RHA1 has been localized to the 1,100-kb linear plasmid pRHL1 (Masai et al. 1997; Takeda et al. 2004). Similarly, the bph gene cluster of TA421 was shown to be plasmid localized (Kosono et al. 1997). The complete nucleotide sequence of the Rhodococcus erythropolis BD2 linear plasmid pBD2, which comprises an ipbA1A2A3A4CB gene cluster for isopropylbenzene degradation, has recently been obtained (Stecker et al. 2003). The similarities of the key enzymes and the regulators of the ipb pathway genes in BD2 and the linear plasmid-encoded bph pathways indicate that the ipb and bph operons have been distributed among Gram-positive organisms via plasmid-mediated horizontal gene transfer. Moreover, the ipb gene cluster was shown to be flanked by a total of 22 ORFs showing significant similarities to insertion sequences, integrases and transposases, indicating that the ipb genes (and possibly also the bph genes) were acquired via transposition events and subsequently distributed among the rhodococci via horizontal transfer.
In strains KF715 (Hayase et al. 1990; Lee et al. 1995), LB400 (Mondello 1989) and KF707 (Furukawa and Miyazaki 1986) the biphenyl catabolic genes are chromosomally encoded. The presence of similar chromosome-localized genes in different strains implies that even the chromosomal bph genes possess, or have used, mechanisms for mobilization in other strains (Nishi et al. 2000). In fact, a 90-kb DNA region, including the bph genes, could be transferred by conjugation from KF715 to P. putida AC30 (Nishi et al. 2000). Transconjugants gained the ability to grow on biphenyl and the bph element was located on the chromosome. The element thus behaves like a conjugative transposon.
Conjugative transposons excise by site-specific recombination, transfer the resulting circular form by conjugation, integrating by recombination between a specific site on this circular form and a site in the host genome (Burrus et al. 2002). As these features are shared with “integrative plasmids” and genomic islands, it was recently proposed to classify them all as “integrative and conjugative elements (ICEs)” and the generic term “genomic island” has been proposed for a wider range of large integrated mobile (or potentially mobile) elements (Toussaint et al. 2003). Thus far, the best characterized genomic island carrying bph catabolic genes is Tn4371, a 55-kb element in Wautersia oxalatica A5 comprising the bphEGF(orf4)A1A2A3A4BCD(orf1)A4 gene cluster (Toussaint et al. 2003). Transfer of Tn4371 to other bacteria was initially observed only after insertion of the element into a plasmid with a broad host range, suggesting that the element was a transposon. However, the element was found to integrate easily into the host chromosome at specific locations (Merlin et al. 1999), and the sequence indicates that the element can be considered as a genomic island. Similar to other genomic islands, Tn4371 shows a mosaic structure of several building blocks, including a tyrosine recombinase that is related to phage integrases (Merlin et al. 1999), and transfer most likely involves a site-specific excision/integration process.
Biphenyl 2,3-dioxygenases
A major interest in the analysis of biphenyl-degrading bacteria is their capacity to transform PCB congeners. Based on the analysis of various biphenyl-degrading isolates, it could be deduced that lower chlorinated congeners are more easily transformed than higher chlorinated congeners, and PCB congeners with chlorines on one aromatic ring only are more easily degraded than those bearing chlorine substituents on both aromatic rings. However, each isolate exhibits a particular activity spectrum with regard to the type and extent of PCB congeners metabolized, with some strains having a narrow spectrum and others, notably strain LB400 (Mondello 1989), being able to transform a broad range of congeners.
To a significant extent, the spectrum of PCB congeners that can be transformed by an organism is determined by the specificity of the biphenyl 2,3-dioxygenase, the enzyme that catalyzes the first step in the upper pathway and which belongs to the toluene/biphenyl family of Rieske non-heme iron oxygenases (Gibson and Parales 2000). Studies on various biphenyl 2,3-dioxygenases have revealed considerable differences in their congener selectivity patterns, as well as their preference for the ring attacked (Erickson and Mondello 1993; Kimura et al. 1997; McKay et al. 1997; Seeger et al. 1999; Zielinski et al. 2002). The α-subunit was found to be crucially responsible for recognition and binding of the substrates, and also for substrate specificity (Furukawa et al. 1993; Kimura et al. 1997). As an example, the biphenyl 2,3-dioxygenases of strains LB400 and KF707 differ in only 20 amino acid residues; however, the two enzymes differ dramatically in substrate specificity and regioselectivity. The LB400 dioxygenase shows broad substrate specificity and transforms PCBs with up to six chlorine atoms (Mondello 1989). Congeners with chlorines at the 2- and 5-position on the ring were readily attacked, whereas activity was poor with congeners with chlorines at both para-positions (Mondello 1989; Arnett et al. 2000). 2,5,2′,5′-Tetrachlorobiphenyl was subject to 3,4-dioxygenation (Haddock et al. 1995) (Fig. 2). A detailed analysis of the dioxygenation products of several di- to pentachlorinated biphenyls formed by this enzyme revealed a complex dependence of the regiospecificity and the yield of dioxygenation on the substitution patterns of both the oxidized and the nonoxidized rings (Seeger et al. 1999). Biphenyl-2,3-dioxygenase attack on 2,2′- or 2,4′-dichlorobiphenyl gave rise to virtually quantitative ortho-dechlorination of these congeners by hydroxylation at the chlorinated carbon 2 and its unsubstituted neighbor (Fig. 2). Elimination of chloride leads directly to 2,3-dihydroxy-chlorobiphenyls and obviates the need for biphenyl-2,3-dihydrodiol-2,3-dehydrogenase for the catabolism of such congeners (Seeger et al. 1995). Ortho-dechlorination was also observed for various other 2-chlorosubstituted biphenyls (Haddock et al. 1995; Seeger et al. 1999). Also 2,2′-difluoro-, 2,2′-dibromo-, 2,2′-dinitro-, and 2,2′-dihydroxybiphenyl were accepted as substrates and transformed to 2′-substituted 2,3-dihydroxybiphenyls with concomitant elimination of the substituent (Seeger et al. 2001). Dibenzofuran and dibenzodioxin, which may be regarded as analogues of doubly ortho-substituted biphenyls or diphenylethers, respectively, were attacked at the “quasi ortho” carbon (the angular position 4a) and its neighbor (Fig. 2). This shows that LB400 biphenyl dioxygenase can catalyze angular dioxygenation, a reaction previously assumed to be restricted to certain specialized dioxygenases involved in dibenzo-p-dioxin and dibenzofuran degradation (Armengaud et al. 1998).

Transformation of selected polychlorinated biphenyl (PCB) congeners and of dibenzo-p-dioxin by biphenyl 2,3-dioxygenases of a Burkholderia sp. LB400 (Haddock et al. 1995; Seeger et al. 1995, 1999), b Pseudomonas pseudoalcaligenes KF707 (Suenaga et al. 2002) and c Phe227Val and Phe377Ala mutants of KF707 dioxygenase (Suenaga et al. 2002). Unstable intermediates are enclosed in brackets
Compared to the LB400-derived biphenyl 2,3-dioxygenase, the enzyme of strain KF707 has a much more restricted substrate range and is not capable of ortho-dechlorination, but catalyzes 5,6-dioxygenation of 2,2′-dichlorobiphenyl with low activity (Suenaga et al. 2002) (Fig. 2). However, it is superior in its activity in transforming 4,4′-dichlorobiphenyl (Erickson and Mondello 1993).
Site-directed mutagenesis and gene shuffling methods were applied to identify biphenyl dioxygenase sequences that determine specificity and to construct enzymes combining the broad congener specificity of strain LB400 with increased activity against congeners oxidized especially by the dioxygenase of strain KF707 (Erickson and Mondello 1993; Kimura et al. 1997; Mondello et al. 1997; Kumamaru et al. 1998; Brühlmann and Chen 1999b; Suenaga et al. 1999, 2002). Positions 335, 376 and 377 (numbering according to KF707 dioxygenase) were identified to be particularly important for specificity. Mutation of Thr376 (KF707) to Asn376 (LB400) in KF707 dioxygenase resulted in an expansion of the range of biotransformable PCB congeners (Kimura et al. 1997). Replacement of Phe335 by Ile in LB400 dioxygenase improved the capacity to transform 4,4′-dichlorobiphenyl. In further attempts to modify the KF707 enzyme it was shown that mutations in position 335 (Ile335Phe), 376 (Thr376Asn) and 377 (Phe377Leu) enabled the enzyme to perform ortho-dechlorination and to transform 2,5,2′,5′-tetrachlorobiphenyl. Phe227Val and Phe377Ala mutants resulted in a completely new ability to meta-dechlorinate 3,3′-dichlorobiphenyl; those variants, however, completely lost the ability to transform other PCB congeners (Suenaga et al. 2002) (Fig. 2). However, the greatest improvements in activity over wild-type enzymes usually resulted from multiple amino acid modifications, suggesting that the effects of these mutations are cooperative (Mondello et al. 1997; Suenaga et al. 2001a). The availability of an enzyme model based on the available crystal structure of a related enzyme [a three-dimensional structure of naphthalene dioxygenase is available (Kauppi et al. 1998)] has helped significantly in the design of directed mutagenesis studies and in explaining the effects obtained by gene shuffling experiments. However, in various cases, residues that are not in the immediate vicinity of the bound substrate were found to significantly influence the structure of the active site (Zielinski et al. 2003). The medium- or long-range interactions responsible for such indirect effects are currently only poorly predictable. Moreover, the structure of the substrate-binding site is determined by segments that are rather distant in the sequence. Thus, the effect of individual exchanges of such segments on binding site structure will vary depending on the structures forced by other segments, and generalization of conclusions drawn from individual effects in single experiments must be treated with caution (Zielinski et al. 2002).
Efforts to create chimeric dioxygenases with novel specificity/regioselectivity are on the one hand forced by the need to optimize PCB transformation efficacy (Barriault et al. 2002). On the other hand, optimized hybrid biphenyl dioxygenases are useful not only for the transformation of trichloroethylene (Maeda et al. 2001) but also for the introduction of hydroxyl groups into flavonoids to produce antioxidants and cancer chemoprotectants (Chun et al. 2003; Kim et al. 2003; Seeger et al. 2003).
Diversity in “upper pathway” enzymes
Although certain PCBs serve as substrates for biphenyl dioxygenases, PCB-degrading organisms do not usually use PCBs as an energy source, but rather catabolize these substrates cometabolically. Therefore, it is not surprising that metabolites of the upper pathway may be formed as dead-end products (Furukawa et al. 1979; Bedard and Haberl 1990; Seeger et al. 1997).
The second step in the metabolic pathway is catalyzed by cis-2,3-dihydro-2,3-dihydroxybiphenyl dehydrogenase. Cis-dihydrodiol dehydrogenases are involved in various aromatic degradation pathways and are usually members of the family of short-chain alcohol dehydrogenases. Generally, cis-dihydrodiol dehydrogenases are broad substrate enzymes, and most dehydrogenases are able to transform several cis-dihydrodiol isomers (Patel and Gibson 1974; Rogers and Gibson 1977; Raschke et al. 1999). Even though the specificity of BphB enzymes for differently chlorinated biphenyl dihydrodiols has not yet been rigorously determined, it was shown that BphB of LB400 dehydrogenates all 2,3-biphenyl dihydrodiols chlorinated at the non-oxidized ring except for 2′,6′-substituted compounds (Hülsmeyer et al. 1998). In addition, various dihydrodiols chlorosubstitued at both aromatic rings were dehydrogenated (Hülsmeyer et al. 1998). Even the 2,2′,5,5′-tetrachlorinated derivative of biphenyl 3,4-dihydrodiol, initially assumed not to be dehydrogenated (Haddock and Gibson 1995), was shown to be transformed by BphB of LB400 as well as by BphB of Comamonas testosteroni B-356 (Barriault et al. 1999). Despite the broad substrate range of BphB, this enzyme was determined as a crucial pathway bottleneck in the transformation of PCBs when combined with a highly active dioxygenase (Brühlmann and Chen 1999a).
The third reaction in the bph pathway is catalyzed by 2,3-dihydroxybiphenyl 1,2-dioxygenases, which are of particular significance in the degradation of PCBs, as they are subject to various types of inhibition including suicide inactivation (Vaillancourt et al. 2002). 2,3-Dihydroxybiphenyl 1,2-dioxygenases belong to the family of extradiol dioxygenases and use mononuclear Fe2+ to cleave dihydroxybiphenyl. Usually, extradiol dioxygenases, which are involved in biphenyl degradation, belong to the I.3.A subfamily of extradiol dioxygenases (Eltis and Bolin 1996) and are specialized for transformation of 2,3-dihydroxybiphenyls. These enzymes differ in their substrate specificity, but seem to be generally capable of transforming various chlorosubstituted derivatives (Hein et al. 1998; Dai et al. 2002). However, both 3,4-dihydroxybiphenyl (Lloyd-Jones et al. 1995; McKay et al. 2003) and 2′-chlorosubstituted 2,3-dihydroxybiphenyls strongly inhibit biphenyl dioxygenases. 2′-Chlorosubstituted 2,3-dihydroxybiphenyls promote suicide inactivation, which involves release of superoxide during catalysis and oxidation of the active site Fe2+, and thus interfere with the degradation of other compounds (Dai et al. 2002). However, a new class of single domain extradiol dioxygenases, thus far observed only in Sphingomonas (Heiss et al. 1995) and Rhodococcus strains (Asturias and Timmis 1993; Sakai et al. 2002), has been shown to have adapted to the transformation of 2-chlorosubstituted 2,3-dihydroxybiphenyls (McKay et al. 2003; Vaillancourt et al. 2003).
The fourth step in the bph pathway is catalyzed by HOPDA hydrolase BphD, which hydrolyzes HOPDA at a carbon-carbon bond to yield 2-hydroxypenta-2,4-dienoate and benzoate. Recent studies using LB400 BphD revealed that this enzyme relatively rapidly transformed HOPDAs bearing chlorine substituents at the phenyl moiety, whereas HOPDAs bearing chlorine substituents on the dienoate moiety were poor substrates, although they act as competitive inhibititors of BphD (Seah et al. 2000). Moreover, HOPDAs undergo nonenzymatic transformation to products that included acetophenones (Seah et al. 2000). These results explain the accumulation of HOPDAs and chloroacetophenones in the microbial degradation of certain PCB congeners. More significantly, they indicate that during the degradation of PCB mixtures, BphD would be inhibited, thereby slowing the mineralization of all congeners. BphD is thus a key determinant in the aerobic microbial degradation of PCBs. Even though the R. globerulus P6-derived BphD also exhibits significantly higher activity with HOPDAs bearing chlorine substituents at the phenyl moiety compared to HOPDAs bearing chlorine substituents at the dienoate moiety, it was shown that these bphD gene products differ significantly in their kinetic properties.
Metabolic complexity in Rhodococcus
A simplified assumption in biphenyl degradation and PCB catabolism is that the strains isolated have a single set of biphenyl degradation genes clustered in a single locus, and that only the corresponding gene products are responsible for the PCB-transforming capability of the strain. In recent years, it has become clear that various microorganisms contain multiple metabolic pathways or isoenzymes involved in PCB metabolism. R. globerulus P6 was shown early to contain at least three BphC-encoding genes (Asturias and Timmis 1993), with two of them being expressed during growth on biphenyl (McKay et al. 2003). Recent analysis indicated that this strain contains at least four BphC genes (Kosono et al. 1997). Six extradiol dioxygenase genes were identified in strain RHA1 (Sakai et al. 2002), and three are expressed when the organism is grown on biphenyl. A total of seven bphC genes were found in strain TA421 (Maeda et al. 1995; Kosono et al. 1997) and eight in R. rhodochrous K37 (Taguchi et al. 2004). Thus, the presence of multiple extradiol dioxygenases seems to be common in rhodococcal strains and is thought to contribute to the versatility of this group of bacteria with respect to the degradation of haloaromatic compounds. Later, it was observed that strain RHA1 also harbors multiple aromatic ring hydroxylating dioxygenases (Kitagawa et al. 2001b) and hydrolases (Yamada et al. 1998), which could function in the transformation of PCB congeners. The complex gene organization (Shimizu et al. 2001; Takeda et al. 2004) resembles slightly that previously observed in a dibenzo-p-dioxin degrading Sphingomonas strain (Armengaud et al. 1998).
Biphenyl pathways in Sphingomonas
Biphenyl-degrading Sphingomonas strains such as Sphingobium yanoikuyae B1 (Gibson et al. 1973; Zylstra and Kim 1997), Sphingobium yanoikuyae Q1 (Furukawa et al. 1983; Taira et al. 1988), Sphingomonas chungbulensis DJ77 (Kim et al. 2000) or Novosphingobium aromaticivorans F199 (Romine et al. 1999) have also been described, and genetic analysis has revealed a highly complex arrangement of catabolic genes differing immensely in sequence homology and gene order from those reported for other genera (Fig. 3). In strain B1, a bphB gene was localized after a transposon mutation rendered the strain incapable of mineralizing biphenyl (Kim and Zylstra 1995). Analysis of a 40 kb DNA fragment comprising this gene revealed the presence of genes for no less than five different oxygenase α subunits (bphA1a-e) (Zylstra and Kim 1997), with only two of them (bphA1a and b) having adjacent genes for the β subunit. However, deletion or inactivation by insertional mutagenesis of any single α-subunit gene does not impair the ability of B1 to oxidize biphenyl, indicating that the biphenyl oxygenase α-subunit-encoding genes are located in a different gene region. In contrast, single genes for ferredoxin (bphA3) and a ferredoxin reductase (bphA4) were found, with lack of the ferredoxin resulting in a nonfunctional biphenyl dioxygenase; growth on m-xylene, phenanthrene and 3-methylbenzoate was also abolished (Kim and Zylstra 1999), implying that the ferredoxin is associated with multiple terminal oxygenases. Complete sequencing of the 184 kb catabolic plasmid pNL1 from strain F199 showed the presence of a gene region with high homology to that identified in B1. Thirty-five genes (Fig. 3) are arranged in the same order and transcriptional direction (Romine et al. 1999) and there is a mixture of genes probably involved in biphenyl catabolism (bph-genes) and those probably involved in xylene/toluene/methylbenzoate (xyl-genes) degradation, including xylJQK genes, which probably fulfill a function in 3-methylbenzoate degradation, but could possibly be recruited for 2-hydroxypenta-2,4-dienoate degradation formed by hydrolysis of HOPDA. As well as five genes encoding an α-subunit, a sixth terminal oxygenase (bphA1fA2f) was identified and suggested to encode the terminal dioxygenase component of biphenyl/naphthalene dioxygenase. However, no evidence for this function, nor the function of predicted bphDEF genes have been proposed thus far. For a more detailed review on aromatic catabolic genes in Sphingomonads, see Pinyakong et al. (2003).

Transformation of biphenyls by enzymes distinct from those of the biphenyl upper pathway
Despite differences in genetic organization, the initial enzymatic steps in the aerobic degradation of many aromatic compounds in different bacterial genera are very similar, and enzymes homologous to BphA, B and C are found in pathways responsible for the degradation of naphthalene, benzoate and benzene (Fig. 4). Despite the evolutionary adaptation of enzymes to specific substrates, the enzymes of a particular pathway usually catalyze the transformation of a range of substrate analogues. Hence, enzymes in the PCB pathways may transform not only PCBs and their metabolites, but also other related compounds such as monocyclic aromatics (Suenaga et al. 2001b), and vice versa. This substrate overlap means that other pollutants in a site may act as co-substrates that can influence the composition and activity of biphenyl-metabolizing bacteria. As an example, chlorobenzene dioxygenase of Pseudomonas sp. P51 (which belongs, like biphenyl dioxygenases, to the toluene/biphenyl family of Rieske non-heme iron oxygenases) (Fig. 4) was shown to be capable of efficiently transforming monochlorinated biphenyls (Raschke et al. 1999, 2001); however, higher substituted biphenyls were not attacked. Even naphthalene dioxygenase of P. putida G7 (belonging to the naphthalene family of Rieske non-heme iron oxygenases) (Fig. 4) can efficiently transform chlorinated biphenyls (Barriault and Sylvestre 1999b). However, this enzyme differed in regioselectivity and a significant amount of biphenyl was subject to 3,4-dioxygenation (Barriault and Sylvestre 1999a; Parales et al. 2000). Because 3,4-dihydroxylated biphenyls were cleaved efficiently by the subfamily I.3.E extradiol dioxygenase DoxG of Pseudomonas sp. strain C18 (Eltis and Bolin 1996), it was suggested to use naphthalene catabolic enzymes to expand the capabilities of the bph pathway to degrade PCBs (Barriault et al. 1998).

Phylogenetic tree of the α-subunits of selected Rieske non-heme iron oxygenases. Families identified by Gibson and Parales (2000) are indicated. For clarity, only protein and strain designations are indicated
Interestingly, not all aromatic ring-hydroxylating dioxygenases are capable of biphenyl transformation and whereas phenanthrene dioxygenase PhnA1A2A3A4 of Cytoclasticus sp. strain A5 exhibited such activity (Kasai et al. 2003), the phenanthrene dioxygenase PhdABCD of Nocardioides sp. strain KP7 (Fig. 4) was not active against biphenyl (Shindo et al. 2001). However, a systematic evaluation of different groups of aromatic ring hydroxylating dioxygenases for their capability to transform PCBs is still lacking. Nevertheless, the capacity to transform biphenyls is evidently not restricted to biphenyl-degrading organisms. Growth on biphenyl even in the absence of a biphenyl pathway has been reported. As an example, introduction of the capacity to mineralize toluene via the tod pathway into P. putida F1 (Zylstra et al. 1988) allowed the strain to mineralize biphenyl (Furukawa et al. 1993), showing that toluene dioxygenase (TodC1C2BA), toluene dihydrodiol dehydrogenase (TodD) and the meta-cleavage enzyme TodE have significant cross-reactivity with biphenyl or metabolites produced during biphenyl degradation, but that TodF 2-hydroxy-6-oxohepta-2,4-dienoate hydrolase cannot deal with HOPDA. Recently, P. putida strain CE2010 was reported to mineralize biphenyl by a similar mosaic of tod (toluene) and cmt (cumate) pathways in the absence of an authentic biphenyl pathway (Ohta et al. 2001). It is possible that such mosaic routes, both within individual strains and within communities, are important for the metabolism of complex pollutant mixtures.
Metabolic pathways for chlorobenzoate degradation
The bph-encoded biphenyl pathway enzymes present in a variety of native bacteria transform many PCBs. However, most of those bacteria only cometabolize chlorinated congeners, and thus, concomitant with the transformation of PCBs, different dead-end metabolites accumulate, including dihydrodiols (Brühlmann and Chen 1999a), dihydroxybiphenyls (including 3,4-dihydroxylated derivatives) (Triska et al. 2004), or chlorinated HOPDAs (Furukawa et al. 1979; Seeger et al. 1997). It was recently shown that dihydrodiols and dihydroxybiphenyls are very toxic metabolites for bacteria, affecting cell viability—and thus bacterial performance—much more than (chloro)biphenyls (Cámara et al. 2004). It will be of critical importance to overcome dead-end reactions in the catabolic process and balance the activities of enzymes involved in the degradation to avoid accumulation of toxic metabolites. However, as different pathway bottlenecks for different PCB congeners operate in different organisms, is it expected that only diverse communities with complementary activities will to able to deal with complex PCB mixtures.
The complexity of PCB degradation is enhanced by the fact that most PCB degraders at best transform chlorinated biphenyls into chlorinated benzoates and thus live at the expense of the 2-hydroxypenta-2,4-dienoate generated during hydrolysis of HOPDAs. Even though chlorobenzoates themselves are obviously non-toxic to bacteria, the negative effects of chlorobenzoate metabolism on chlorobiphenyl degradation have already been reported (Havel and Reineke 1992; Sondossi et al. 1992; Stratford et al. 1996) and thus the metabolic fate of chlorobenzoates must be understood when analyzing PCB metabolism.
Transformation of chlorobenzoates by benzoate degradative pathways
Benzoate 1,2-dioxygenase genes are widespread and have been identified in a wide variety of microorganisms and localized in diverse genome sequencing projects. The substrate specificity of benzoate dioxygenases is usually restricted to benzoate and 3-chloro-/3-methylbenzoate, as shown for chromosomally encoded enzymes from strain Pseudomonas sp. B13 (Reineke and Knackmuss 1978a), and Wautersia eutropha JMP134 (Pieper et al. 1993), as well as for the distantly related enzyme from Rhodococcus sp. strain RHA1 (Kitagawa et al. 2001a). Enzymes from both B13 and JMP134 form 3-chloro- and 5-chlorodihydrodihydroxybenzoate in a 2:1 ratio (Reineke and Knackmuss 1978a), which are dehydrogenated to 3-chloro- and 4-chlorocatechol, respectively (Reineke and Knackmuss 1978b). Chlorocatechols can be regarded as environmentally important intermediates. Their metabolic fate, when processed by enzymes of widespread pathways for catechol metabolism, can be of environmental significance.
The 3-oxoadipate pathway
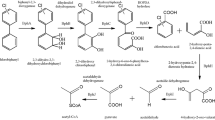
The chromosomally encoded 3-oxoadipate pathway is widely distributed in soil bacteria and fungi (Harwood and Parales 1996). One branch of this pathway converts protocatechuate, whereas the other converts catechol. Two additional steps, a 3-oxoadipate:succinyl-CoA transferase and a 3-oxoadipyl-CoA thiolase, accomplish the conversion of 3-oxoadipate to tricarboxylic acid cycle intermediates (Stanier and Ornston 1973). Enzyme studies and amino acid sequence data indicate that the pathway is highly conserved in diverse bacteria. In the catechol branch, catechol is cleaved by catechol 1,2-dioxygenases giving rise to muconate. Usually, catechol 1,2-dioxygenase transforms 4-chlorocatechol with relatively high activity (Dorn and Knackmuss 1978a; Schmidt et al. 1980; Blasco et al. 1997). Muconate cycloisomerases catalyze dehalogenation and decarboxylation of 3-chloromuconate, the product of 4-chlorocatechol ring-cleavage, to form protoanemonin (Blasco et al. 1995) (Fig. 5), a compound of high toxicity (Seegal and Holden 1945). Protoanemonin formation in turn was assumed to be the reason for the poor survival of polychlorobiphenyl cometabolizing organisms in soil microcosms due to channeling of intermediary 4-chlorocatechol into the 3-oxoadipate pathway (Blasco et al. 1997). Chloroprotoanemonin was even reported to be formed from 2,4-dichloromuconate by muconate cycloisomerase (Kaulmann et al. 2001). In the case of 2-chloromuconate turnover, muconate cycloisomerases catalyze cycloisomerization only, to form both 2-chloro- and 5-chloromuconolactone as stable products (Vollmer et al. 1994) (Fig. 5). Both chloromuconolactones are substrates of proteobacterial muconolactone isomerases (Prucha et al. 1996a, b; Skiba et al. 2002) of the 3-oxoadipate pathway. Muconolactone isomerase catalyzes dehalogenation of 5-chloromuconolactone to form predominantly cis-dienelactone (Fig. 5). The proposed mechanism acts via abstraction of the C4 proton followed by spontaneous chloride elimination. 2-Chloromuconolactone was transformed into protoanemonin (Fig. 5), probably by elimination of CO2 and chloride from chlorosubstituted 3-oxoadipate enol-lactone (Skiba et al. 2002). Thus, the presence of a 3-oxoadipate pathway will severely interfere with the degradation of PCBs via chlorobenzoates as intermediates.

Diversity of pathways for the degradation/transformation of chlorocatechols. C230 Catechol 2,3-dioxygenase, CC23O chlorocatechol 2,3-dioxygenase, C12O catechol 1,2-dioxygenase, CC12O chlorocatechol 1,2-dioxygenase, MCI muconate cycloisomerase, CMCIP proteobacterial chloromuconate cycloisomerase, CMCIR chloromuconate cycloisomerase of R. opacus 1CP, MLI muconolactone isomerase, CMLIR chloromuconolactone isomerase of R. opacus 1CP, DLHP proteobacterial dienelactone hydrolase, DLHR dienelactone hydrolase of R. opacus 1CP, tDLH trans-dienelactone hydrolase, MAR maleylacetate reductase. Unstable intermediates are enclosed in brackets
The meta-cleavage pathways
Catechol meta-cleavage routes are also widespread and are usually involved in the degradation of methylsubstituted compounds such as toluene or methylphenols (Bayly et al. 1966; Worsey and Williams 1975; Zylstra et al. 1988; Shingler et al. 1992). For a long time the presence of a catechol meta-cleavage pathway was generally assumed to severely interfere with the degradation of chloroaromatics. One of the reasons for this interference was assumed to be the formation of a suicide product, reactive acyl chloride from 3-chlorocatechol (Fig. 5), resulting in irreversible inactivation of the catechol 2,3-dioxygenase of P. putida mt-2 (Bartels et al. 1984). However, in other cases reversible inactivation was shown to be due to rapid oxidation of the active site ferrous iron into its ferric form with concomitant loss of activity (Vaillancourt et al. 2002), and a general mechanism for the inactivation of extradiol dioxygenases during catalytic turnover involving the dissociation of superoxide from the enzyme-catecholic-dioxygen ternary complex was suggested. However, independent of the inactivation mechanism involved, a general inhibitory effect of 3-chlorocatechol formed during the metabolism of 3-chlorobiphenyl on biphenyl metabolism was observed, at least in C. testosteroni B-356 (Sondossi et al. 1992), most probably caused by inactivation of BphC.
In contrast to 3-chlorocatechol, 4-chlorocatechol is a moderate substrate for various catechol 2,3-dioxygenases (Murray et al. 1972; Bartels et al. 1984; Kitayama et al. 1996), among them catechol 2,3-dioxygenases of family I.2.A (Eltis and Bolin 1996). However, despite high sequence identity, members of this subfamily exhibit very different substrate specificities (Kitayama et al. 1996). Some authors postulated that compounds degraded via 4-chlorocatechol might be mineralized via a catechol-degrading meta-cleavage pathway (Janke and Fritsche 1979; Higson and Focht 1992; Arensdorf and Focht 1994; Hollender et al. 1994, 1997; McCullar et al. 1994; Arensdorf and Focht 1995). B. cepacia P166 was shown to mineralize 4-chlorobiphenyl via 4-chlorobenzoate, 4-chlorocatechol followed by a meta-cleavage route (Arensdorf and Focht 1995). 3-Chlorocatechol metabolism, in contrast, was toxic to the strain (Arensdorf and Focht 1994).
Mineralization of chlorobenzoates
In recent years, it has become more and more evident that bacteria have evolved various strategies to degrade chlorobenzoates, and degradation can occur via chlorocatechol (clc pathway and others) (Dorn and Knackmuss 1978b), via hydrolytic dehalogenation of 4-chlorobenzoate to give 4-hydroxybenzoate [fcb pathway (Klages and Lingens 1979)], via 4,5-dioxygenation of 3-chlorobenzoate and 3,4-dichlorobenzoate to give 5-chloroprotocatechuate (Nakatsu and Wyndham 1993), and probably via a fourth pathway with gentisate as intermediate [gp-pathway (Krooneman et al. 1996, 1998)].
The chlorocatechol pathway
In most organisms isolated thus far, chlorocatechols are degraded by a chlorocatechol ortho-cleavage pathway (Dorn and Knackmuss 1978a, b; Schmidt and Knackmuss 1980; Schmidt et al. 1980; Kaschabek and Reineke 1992), where degradation is initiated by a broad specificity chlorocatechol 1,2-dioxygenase to produce the corresponding chloro-cis,cis-muconates (Dorn and Knackmuss 1978a, b; Schmidt et al. 1980; Broderick and O’Halloran 1991) (Fig. 5). As chlorocatechols are highly toxic (Schweigert et al. 2001), microorganisms grow on chloroaromatics only when they possess an activity equilibrium between chlorocatechol-producing and -consuming activities (Klemba et al. 2000; Perez-Pantoja et al. 2003).
A key enzyme in the pathway is chloromuconate cycloisomerase, which differs in various aspects from muconate cycloisomerases (Fig. 5). Proteobacterial chloromuconate cycloisomerases catalyze a specific cycloisomerization of 2-chloromuconates to form preferentially 5-chloromuconolactone, which is dehalogenated to trans-dienelactone (Vollmer and Schlömann 1995). Thus, proteobacterial chloromuconate cycloisomerases are specific dehalogenases. The formation of trans-dienelactone is due to the fact that, after cycloisomerization, the lactone ring has to rotate in the catalytic center to achieve proton abstraction and thus dehalogenation (Schell et al. 1999). In contrast, chloride from the 3-position of chloromuconate seems to be directly eliminated during cycloisomerization (Kaulmann et al. 2001) to form cis-dienelactone. Evidently, for the degradation of various chloroaromatics, an important prerequisite is that the dienelactone hydrolase involved in the pathway is of relaxed substrate specificity and accepts both cis- and trans-isomers of dienelactone as substrates (Schlömann et al. 1990). The maleylacetates formed are then transformed by a maleylacetate reductase, which catalyzes the reduction of the double bond to form 3-oxoadipate (Kaschabek and Reineke 1992). Maleylacetates with chlorine substituents in the 2-position, such as 2-chloromaleylacetate (formed from 3,5-dichloro-, and 3,6-dichlorocatechol), are reduced in a first step to give maleylacetate. Obviously, enzymatic attack on the C2-carbon results in an intermediate that spontaneously eliminates chloride.
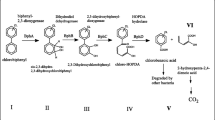
The genes encoding chlorocatechol pathway enzymes form clusters, and the structures of the corresponding operons are highly conserved, in spite of the geographically distinct origins of the bacteria or differences in their phylogenetic position (Fig. 6). In P. putida AC858, Pseudomonas sp. strain P51 (van der Meer et al. 1991) and others, the gene cluster encoding chlorocatechol dioxygenase, chloromuconate cycloisomerase, dienelactone hydrolase and maleylacetate contain an ORF of unknown function following the chloromuconate cycloisomerase gene. Such an ORF is absent in the otherwise identically structured chlorocatechol gene clusters tfdI of Wautersia eutropha JMP134 (Don and Pemberton 1981, 1985), and tfd of Burkholderia sp. strain NK8 (Liu et al. 2001). Regulatory genes orientated in the opposite direction precede the catabolic genes; their gene products, members of the LysR family, act as positive regulators (Coco et al. 1993) with chloromuconates as effectors (McFall et al. 1997; Ogawa et al. 1999). 2-Chloro- and 3-chlorobenzoate, as well as 3-chloro- and 4-chlorocatechol, were shown to act as effectors of the TfdT regulator of Burkholderia sp. strain NK8 (Liu et al. 2001). In contrast to organisms isolated based on their capacity to degrade chlorobenzoates or chlorobenzenes, organisms capable of degrading 2,4-dichlorophenoxyacetate usually contain a tfdRCEBKA gene cluster, comprising a chlorocatechol dioxygenase and dienelactone hydrolase-encoding gene together with those encoding a chlorophenol hydroxylase, a transport protein and a 2,4-D/α-ketoglutarate dioxygenase, but lack the tfdD and tfdF genes encoding chloromuconate cycloisomerases and maleylacetate reductase encoding genes (Vedler et al. 2000; Poh et al. 2002; Hoffmann et al. 2003) (Fig. 6). But not only the structures of the chlorocatechol gene are conserved: isofunctional enzymes of the chlorocatechol pathway were shown to be homologous and more similar to each other than to the corresponding enzymes of catechol pathways, indicating a common origin of the proteobacterial chlorocatechol pathways (Schlömann 1994). In contrast, the enzymes of the Gram-positive organism Rhodococcus opacus 1CP turned out to have unusual biochemical properties and sequences with relatively little similarity to proteobacterial chlorocatechol gene sequences, thus indicating an independent origin (Eulberg et al. 1998; Moiseeva et al. 2002; Solyanikova et al. 1995).

Genetic organization of chlorocatechol gene clusters clcR-ABCDE in P. putida AC858 (Chatterjee et al. 1981b) [similar gene clusters were observed in P. aeruginosa JB2, clcR-ABCDE (Hickey et al. 2001); Pseudomonas sp. P51, tcbR-CD ORF EF (van der Meer et al. 1991); P. chlororaphis RW71, tetR-CD ORF EF (Potrawfke et al. 2001) and W. eutropha NH9, cbnR-ABCD, (Ogawa and Miyashita 1999)]; tfdT-CDEF in Burkholderia sp. NK8 (Liu et al. 2001), tfdA-S—R-D II C II E II F II B II K— T-C I D I E I F I B I in W. eutropha JMP134 (Trefault et al. 2004); tfdD—R-CEBKA—F in Burkholderia cepacia 2a (pIJB1) (Poh et al. 2002) [similar gene clusters were observed in Variovorax paradoxus TV1 (pTV1) (AF028643) and Achromobacter xylosooxidans subsp. denitrificans EST4002 (pEST4011) (Vedler et al. 2000)], tfdR-C II E II BKA—FE-DC-R in Deftia acidovorans P4a (Hoffmann et al. 2003), and clcB—R-AD and clcA2D2B2F in Rhodococcus opacus 1CP (Moiseeva et al. 2002)
Mating methods mimicking natural gene transfer were used to transfer genes for chlorocatechol degradation (clc) from strain B13 to various recipients (Reineke and Knackmuss 1979; Reineke 1998). It had been suspected that the genes were transferred by means of a conjugative plasmid, pB13 (Chatterjee and Chakrabarty 1983). However, in contrast to various other chlorocatechol genes, which are obviously localized on plasmids (Chatterjee et al. 1981a; van der Meer et al. 1991; Trefault et al. 2004), the clc genes of B13 reside on a 105 kb conjugative genomic island (the clc element) in two copies on the chromosome (Ravatn et al. 1998a, b). In contrast to many pathogenicity islands, the clc element is self-transmissible to other recipient bacteria (Ravatn et al. 1998c; Springael et al. 2002). With a certain frequency the element excises, resulting in a circular intermediate that can be transferred to a new recipient. It integrates into the 3′-end of a target glycine tRNA gene and employs a phage-type integrase, IntB13, to achieve site-specific integration (Ravatn et al. 1998b; van der Meer et al. 2001). Integrase expression was significantly stimulated by growing cultures on 3-chlorobenzoate (Sentchilo et al. 2003), indicating that this chloroaromatic stimulates transfer of the genes encoding its own metabolism.
Alternative pathway of 3-chlorocatechol degradation in Rhodococcus opacus 1CP
Among Gram-positive organisms, chlorocatechol catabolism has been investigated mainly in R. opacus 1CP. 4-Chloro- and 3,5-dichlorocatechol are degraded by an enzymatic sequence similar to that in proteobacteria (Solyanikova et al. 1995; Eulberg et al. 1998); however, the enzymes involved differ in various aspects from those described in proteobacteria (Maltseva et al. 1994a). Pronounced differences were observed between proteobacterial chloromuconate cycloisomerase and the Rhodococcus enzyme, as the latter showed high specificity constants for 3-chloro- and 2,4-dichloromuconate (Maltseva et al. 1994a), but negligible activity with 2-chloromuconate. Moreover, 2-chloromuconate is not dehalogenated but transformed to 5-chloromuconolactone only, whereas 3-chloro- and 2,4-dichloro-cis,cis-muconate are converted to the expected (2-chloro-)cis-dienelactone metabolites (Fig. 5). The Rhodococcus dienelactone hydrolase is special as this enzyme is practically inactive against trans-dienelactone (Maltseva et al. 1994b). The sequence of the Rhodococcus 4-chlorocatechol/3,5-dichlorocatechol catabolic gene cluster reveals a low sequence similarity to the corresponding proteobacterial genes and, together with biochemical evidence, suggests that the chlorocatechol pathway of strain 1CP should have evolved to be functionally convergent to the pathway in proteobacteria (Maltseva et al. 1994b). After adaptation, the strain grew on chloroaromatics, which are metabolized via 3-chlorocatechol (Moiseeva et al. 1999), and a second chlorocatechol 1,2-dioxygenase of broad substrate specificity, a second chloromuconate cycloisomerase, and a second dienelactone hydrolase were identified (Moiseeva et al. 2001). The second chloromuconate cycloisomerase is specific for 2-chloro-cis,cis-muconate; however, in contrast to proteobacterial chloromuconate cycloisomerases, it cannot catalyze dehalogenation, and 5-chloromuconolactone was observed as product (Moiseeva et al. 2001, 2002). Dehalogenation is achieved by an enzyme with high sequence similarity to the muconolactone isomerases of the 3-oxoadipate pathway (Fig. 5), which in proteobacteria have been shown to be capable of dehalogenating 5-chloromuconolactone to cis-dienelactone. Transformation of cis-dienelactone in the pathway is achieved by a second dienelactone hydrolase, which, like its counterpart in 4-chlorocatechol degradation, is specific for cis-dienelactone. A major difference between chlorocatechol degradation in proteobacteria and in Rhodococcus is based on the fact that cis-dienelactone is the intermediate of both 4-chloro- and 3-chlorocatechol degradation in Rhodococcus, whereas proteobacteria form cis- and trans-dienelactone, respectively, thus requiring a broad substrate specificity dienelactone hydrolase.
Alternative ortho-cleavage pathway of 4-halocatechol degradation
As muconate cycloisomerases form protoanemonin as a toxic dead-end product (Blasco et al. 1995), transformation of 4-chlorocatechol by enzymes of the 3-oxoadipate pathway constitutes a problematic catabolic misrouting in bacterial communities (Blasco et al. 1997). A new strategy for avoiding protoanemonin formation has been elucidated in Pseudomonas sp. strain MT1 (Pelz et al. 1999). Strain MT1 degrades 4-chlorocatechol without having a chlorocatechol pathway and a trans-dienelactone hydrolase was shown to be the key enzyme of a novel metabolic route (Nikodem et al. 2003). 4-Chlorocatechol is subject to ring-cleavage by a catechol 1,2-dioxygenase to yield 3-chloromuconate. 3-Chloromuconate is transformed into protoanemonin as predominant reaction product by muconate cycloisomerase of strain MT1, as previously shown for other muconate cycloisomerases (Fig. 5). Protoanemonin is obviously a dead-end product of the pathway. However, a trans-dienelactone hydrolase acts on 4-chloromuconolactone as an intermediate in the muconate cycloisomerase-catalyzed transformation of 3-chloromuconate, thus preventing protoanemonin formation in favor of maleylacetate formation (Fig. 5). The maleylacetate thereby formed is reduced by maleylacetate reductase. Chlorocatechol degradation in strain MT1 thus occurs via a new pathway consisting of a patchwork of reactions known from the 3-oxoadipate pathway (catechol 1,2-dioxygenase, muconate cycloisomerase), the chlorocatechol pathway (maleylacetate reductase) and a trans-dienelactone hydrolase.
Metabolism of 3-chlorocatechol by the chlorocatechol meta-cleavage pathway
Recently, P. putida GJ31 was found to degrade chlorobenzene rapidly via 3-chlorocatechol using a meta-cleavage pathway (Mars et al. 1997). In contrast to other catechol 2,3-dioxygenases, which are subject to rapid inactivation, the chlorocatechol 2,3-dioxygenase of strain GJ31 productively converts 3-chlorocatechol (Kaschabek et al. 1998; Mars et al. 1999). Stoichiometric displacement of chloride leads to the production of 2-hydroxymuconate (Fig. 5), which is further converted through the meta-cleavage pathway. Additional pseudomonads using a meta-cleavage route for 3-chlorocatechol degradation were recently isolated from various contaminated environments (Göbel et al. 2004) suggesting that productive meta-cleavage of 3-chlorocatechol is not atypical for chloroaromatic degradation. The new isolates, Pseudomonas fluorescens SK1, Pseudomonas veronii 16-6A and Pseudomonas sp. strain MG61, harbor chlorocatechol 2,3-dioxygenases, highly resistant to inactivation during 3-chlorocatechol turnover, sharing 97 % identical amino acids with the enzyme from strain GJ31, thus forming a distinct subfamily of catechol 2,3-dioxygenases.
Microbial degradation of 2-chlorobenzoates
Since the 1980s, different groups have succeeded in isolating bacteria capable of degrading 2-chlorobenzoate. The isolates could be grouped into those capable of degrading 2-chlorobenzoate (Zaitsev and Karasevich 1984; Engesser and Schulte 1989; Fetzner et al. 1989; Higson and Focht 1990; Suzuki et al. 2001) and those also capable of degrading either 2,4-dichloro- or 2,5-dichlorobenzoate (Hickey and Focht 1990; Hernandez et al. 1991; Kozlovsky et al. 1993; Romanov et al. 1993; Pavlu et al. 1999). All these organisms catalyze a 1,2-dioxygenation of 2-chlorobenzoate such that one of the vic-hydroxyl groups in the cis-dihydrodiol is bound to the same carbon as the chloro-substituent (Fig. 7). From such an unstable vic-dihydrodiol, the chloro-substituent will be spontaneously eliminated to form catechol (from 2-chlorobenzoate) or 4-chlorocatechol (from 2,4-dichloro- and 2,5-dichlorobenzoate), respectively. As the degradation of 4-chlorocatechol requires the presence of a chlorocatechol degradative pathway, a preliminary assumption is that organisms capable of degrading 2-chlorobenzoate only, and those capable of also degrading 2,4-dichloro- or 2,5-dichlorobenzoate, differ by the presence of a chlorocatechol pathway. However, two distinct 2-chlorobenzoate-degrading dioxygenase systems have been described. The plasmid-borne two-component 2-halobenzoate 1,2-dioxygenases (oxygenase consisting of α- and β-subunits and a reductase) from strain 2CBS (Fetzner et al. 1992; Haak et al. 1995) and TH2 (Suzuki et al. 2001), are similar to two component toluate (Harayama et al. 1991) and benzoate 1,2-dioxygenases (Cowles et al. 2000) of the family of benzoate dioxygenases (Gibson and Parales 2000) (Fig. 4). These 2-halobenzoate dioxygenases are characterized by their high activity against 2-halosubstituted benzoates (Fetzner et al. 1992; Suzuki et al. 2001) but have negligible activity with 4-chloro-, 2,4-dichloro- or 2,5-dichlorobenzoate. In contrast, the 2-chlorobenzoate dioxygenase system of P. aeruginosa strain 142 is a three-component dioxygenase system (oxygenase consisting of α- and β-subunits, ferredoxin and reductase) (Romanov and Hausinger 1994). Moreover, the α-subunits of 2-chlorobenzoate dioxygenases of strains JB2 (Hickey and Sabat 2001) and 142 (Tsoi et al. 1999) exhibit only 22% sequence identity with that of strain 2CBS, but a significant similarity (42%) to salicylate 5-hydroxylase NagG from Pseudomonas sp. strain U2 (Fuenmayor et al. 1998) (Fig. 4) and putative biphenyl dioxygenase α-subunits from S. aromaticivorans F199 (Romine et al. 1999). Thus, 2-chlorobenzoate dioxygenases are functionally similar, but represent two different lineages with distinct activities.

Pathways for the degradation of chlorobenzoates. Initial metabolic steps of a 3- and 4-chlorobenzoate degradation involving benzoate or toluate 1,2-dioxygenase, b 2-chloro- and 2,5-dichlorobenzoate involving 2-halobenzoate 1,2-dioxygenase, c 4-chlorobenzoate involving hydrolytic dehalogenation, and d 3-chloro- and 3,4-dichlorobenzoate involving 4,5-dioxygenation are shown. Unstable intermediates are enclosed in brackets
The 4-chlorobenzoate hydrolytic pathway
The degradation of 4-chlorobenzoate by a pathway involving an early dehalogenation event, was indicated as early as 1976 (Ruisinger et al. 1976). Since then, various strains of different genera degrading 4-chlorobenzoate by a similar mechanism have been isolated (Klages and Lingens 1979, 1980). The 4-chlorobenzoate dehalogenase system has been shown to consist of three distinct enzymes (Elsner et al. 1991) (Fig. 7). The activation of the substrate is carried out by an ATP-dependent 4-halobenzoate-coenzyme A ligase (Löffler and Müller 1991; Chang et al. 1992; Löffler et al. 1992), which shares significant sequence similarity with proteins catalyzing similar chemistry in the β-oxidation pathway (Babbitt et al. 1992). Dehalogenation is catalyzed by a 4-halobenzoyl-CoA dehalogenase, forming 4-hydroxybenzoyl-CoA (Liang et al. 1993; Löffler et al. 1995) in a hydrolytic substitution reaction. Substrate binding to the dehalogenase active site is followed by nucleophilic attack of the carboxylate side chain of Asp145 at the C4 of the benzoyl group (Yang et al. 1994; Crooks et al. 1995; Liu et al. 1995) to form a Meisenheimer intermediate (Dong et al. 2002). The chloride is displaced from this intermediate because of rearomatization of the benzoyl-group to form an enzyme-ester; hydrolysis of the ester generates the 4-hydroxybenzoyl-CoA product. The last step in the reaction to form 4-hydroxybenzoate is carried out by 4-hydroxybenzoate:coenzyme A thioesterase. The absence of serine, cysteine, or histidine catalytic residues distinguish this protein from all other thioesterases characterized to date (Babbitt et al. 1992; Benning et al. 1998). However, crystallographic investigation revealed a high similarity of its three-dimensional structure to that of β-hydroxydecanoyl thiol ester dehydrase from Escherichia coli (Leesong et al. 1996) and, by superimposing the two structures, it has been possible to identify the putative active site region and to propose a catalytic mechanism whereby the carboxylate side chain of Asp17 activates a water molecule for subsequent nucleophilic attack at the thioester carbonyl carbon of the substrate (Benning et al. 1998).
The 3-chlorobenzoate 4,5-dioxygenase pathway
Another chlorobenzoate transforming enzyme system has been described in Comamonas testosteroni strain BR60 (Providenti and Wyndham 2001). 3-Chlorobenzoate is predominantly subject to 4,5-dioxygenation by CbaAB (3-chlorobenzoate 4,5-dioxygenase and reductase) (Nakatsu et al. 1995) to give 5-chloroprotocatechuate after CbaC-mediated dehydrogenation (Fig. 7). A minor amount of 3-chlorobenzoate is 3,4-dioxygenated, resulting in an unstable dihydrodiol, which spontaneously eliminates chloride to form protocatechuate (Nakatsu et al. 1997). 3,4-Dichlorobenzoate is exclusively dechlorinated to give 5-chloroprotocatechuate (Nakatsu and Wyndham 1993; Nakatsu et al. 1997). The oxygenase belongs to the phthalate family of oxygenases (Gibson and Parales 2000) (Fig. 4), comprising both mono- and dioxygenases, where the oxygenase subunit has an α n subunit configuration (in contrast to other oxygenase systems, which consist of α- and β-subunits). 5-Chloroprotocatechuate serves as substrate for the protocatechuate 4,5-dioxygenase pathway in BR60. As described by Kersten et al. (1982, 1985), 2-pyrone-4,6-dicarboxylate is formed after ring-cleavage by nucleophilic displacement of a halide ion from 5-halosubstituted protocatechuates. Hydrolysis of the pyrone is followed by degradation through this meta-cleavage pathway.
Optimizing PCB degradation under environmental conditions
To overcome limitations due to poisoning caused by the production of toxic metabolites, the application of co-cultures and the construction of hybrid strains have been considered. Microorganisms capable of mineralizing some lower chlorinated biphenyls have been obtained by judicious combination of pathway segments comprising the biphenyl upper pathway, a benzoate/toluate or 2-chlorobenzoate dioxygenase system and a chlorocatechol pathway (Mokross et al. 1990; Havel and Reineke 1991; Hickey et al. 1992; McCullar et al. 1994), usually by in vivo conjugative mating of appropriate strains. Among these, the hybrids B. cepacia JHR22 and Pseudomonas sp. UCR2 have been reported to grow on the environmentally important 2-, 2,4- and 2,5-substituted PCB congeners (Havel and Reineke 1991; Hickey et al. 1992). Use of Pseudomonas-derived chlorobenzoate dehalogenase genes like 4-chlorobenzoate dehalogenase or 2-chlorobenzoate dioxygenase resulted in highly efficient mineralization pathways, due primarily to circumvention of the formation of toxic chlorocatechols (Hrywna et al. 1999; Rodrigues et al. 2001). Actually, the use of hybrid organisms (Havel and Reineke 1993), or coinoculation with chlorobenzoate-degrading organisms could, to a certain extent, enhance PCB mineralization in environmental model systems (Hickey et al. 1993; Blasco et al. 1997).
Degradation in the rhizosphere
Microbial degradation of PCBs in the environment is influenced by various biological, chemical and physical factors as well as the survival of microorganisms in cases where bioaugmentation is the application of choice (Blumenroth and Wagner-Döbler 1998; Barriault et al. 1999; Ahn et al. 2001), and by the fact that higher chlorinated PCBs provide the organisms neither with energy nor with carbon to support the degradation process. The challenge for successful bioremediation of PCB-contaminated soil is to devise methods to encourage the growth of selected microorganisms that are capable of transforming PCBs.
It was speculated that certain plant-derived compounds, such as flavonoids, may serve as growth substrates for PCB-degrading bacteria and may induce bph genes. Donelly et al. (1994) showed a high PCB turnover capacity of biphenyl-degrading organisms after growth with certain flavonoids, suggesting that plant roots could serve as a natural injection system capable of dispensing cometabolites into the rhizosphere and inducing PCB degradation in indigenous microorganisms over long periods. Focht (1995) proposed that plant terpenes might stimulate PCB oxidation and, out of several terpenoid compounds, carvone, limonene and p-cymene induce PCB transformation activity in a biphenyl-degrading Arthrobacter strain (Gilbert and Crowley 1997), explaining why growth in a medium containing Mentha spicata (spearmint containing significant amounts of carvone) resulted in enhanced PCB metabolizing activity of that strain. Similar results were obtained with a PCB-transforming P. stutzeri isolate (Tandlich et al. 2001). Interestingly, the addition of orange peel, ivy leaves, pine needles or eucalyptus leaves to PCB-contaminated soil resulted not only in an enhanced PCB transformation activity, but also in significantly enhanced levels of biphenyl degraders compared to unamended soil (Hernandez et al. 1997), and biphenyl degraders isolated during such experiments could grow on terpenes. However, a clear relationship between terpene degradation and the ability to transform PCBs remains to be elucidated. Nevertheless, root exudates may maintain or induce expression of genes involved in PCB transformation.
As the majority of the microbial population found in the soil is associated with plant roots, the rhizosphere is the ideal site in which to modify microbial populations. Pseudomonas fluorescens F113, an important biocontrol strain for sugar beet, was chosen as host strain for bph genes (Brazil et al. 1995). The modified organism colonized roots as effectively as the wild-type and bph genes were expressed in situ, indicating considerable potential for the manipulation of the rhizosphere as a useful strategy for bioremediation. In a complementary approach, it was shown that phenylpropanoids constitute 84% of the secondary metabolites exuded by Arabidopsis roots (Narasimhan et al. 2003). Comparison in a soil/plant system of a Pseudomonas strain that could efficiently colonize Arabidopsis roots, uses phenylpropanoids and is capable of transforming PCBs, with a mutant incapable of degrading phenylpropanoids showed significantly enhanced PCB degradation by the wild type.
Influence of surfactants on PCB degradation
PCBs are superhydrophobic, have extremely poor solubility in water and are poorly bioavailable. Microbes that utilize hydrophobic substrates often produce surface active substances, and the natural roles of biosurfactants have been claimed to increase the surface area of hydrophobic, water-insoluble growth substrates, increasing their bioavailability by increasing the apparent solubility or desorbing them from surfaces and regulating attachment and detachment of microorganisms to and from surfaces (Ron and Rosenberg 2001). A number of studies on the influence of surfactants on the biodegradation efficacy of PCBs have been made, including chemically produced surfactants (Billingsley et al. 1999), humic substances (Fava and Piccolo 2002) and biosurfactants such lipopeptides (Golyshin et al. 1999) or maltotriose esters (Ferrer et al. 2003). Whereas in some cases an increase in degradation rate was observed after addition of surfactants, in others, surfactants decreased degradation rates. On the one hand, addition of surfactants can provoke changes in community composition resulting in a decrease in the degrader population (Colores et al. 2000). On the other hand, as the addition of surfactants reduces bacterial adhesion to surfaces (Stelmack et al. 1999), the net effect of a surfactant on biodegradation depends on the benefits that result from enhanced solubility of target compounds versus the reduction in direct adhesion of bacteria to those compounds. However, how PCBs are degraded by microbial communities under environmental conditions is still poorly understood.
In attempts to study de novo development of PCB-degrading microbial communities, the formation of composite biofilms consisting of bacteria and clay minerals, in which the clay leaflets were arranged around the bacteria to form hutch-like structures, was observed (Lünsdorf et al. 2000). Obviously, the clay minerals performed an essential role in biofilm formation, probably acting as a nutrient shuttle, mediating transfer of PCBs in a palatable form to the bacteria, and it was suggested that clay minerals might fulfil a similar function in soil. Thus, the contrasting effects of surfactant application are a result of the poorly understood complexity of interactions between soil/sediment, pollutant, surfactant and microorganisms in different environments.
Concluding remarks
Significant advances have been made in recent years in the elucidation of the genetic and biochemical basis of aerobic degradation of PCBs, and detailed knowledge of the enzymatic steps and substrate specificity determinants of key enzymes as well as of different catabolic pathway segments necessary for mineralization is available. Although various approaches have been followed to optimize PCB degradation, e.g., by optimizing key enzymes or combination of pathway segments, it is obvious that such approaches are necessarily limited. Given the facts that (1) PCBs in the environment are complex congener mixtures, (2) different catabolic key enzymes have been identified as bottlenecks for each congener, and (3) optimization of a biocatalyst for a single congener often results in decreasing its performance on other congeners, it becomes increasingly evident that micobial communities have to become the focus of research. In contrast to single bacterial species, which are very unlikely to be able to deal with complex congener mixtures, microbial communities can be more versatile, and carbon sharing through complex metabolic interaction can increase PCB degradation. Gaining an understanding of PCB degradation will require integration of single organism studies with efforts to understand functioning of complex communities.
References
Ahmed M, Focht DD (1973a) Degradation of polychlorinated biphenyls by two species of Achromobacter. Can J Microbiol 19:47–52
Ahmed M, Focht DD (1973b) Oxidation of polychlorinated biphenyls by Achromobacter pCB. Bull Environ Contam Toxicol 10:70–72
Ahn YB, Beaudette LA, Lee H, Trevors JT (2001) Survival of a GFP-labeled polychlorinated biphenyl degrading psychrotolerant Pseudomonas spp. in 4 and 22 degrees C soil microcosms. Microbial Ecol 42:614–623
Aoki Y (2001) Polychlorinated biphenyls, polychlorinated dibenzo-p-dioxins, and polychlorinated dibenzofurans as endocrine disrupters—what we have learned from Yusho disease. Environ Res 86:2–11
Arai H, Kosono S, Taguchi K, Maeda M, Song E, Fuji F, Chung SY, Kudo T (1998) Two sets of biphenyl and PCB degradation genes on a linear plasmid in Rhodococcus erythropolis TA421. J Ferment Bioeng 86:595–599
Arensdorf JJ, Focht DD (1994) Formation of chlorocatechol meta cleavage products by a Pseudomonad during metabolism of monochlorobiphenyls. Appl Environ Microbiol 60:2884–2889
Arensdorf JJ, Focht DD (1995) A meta cleavage pathway for 4-chlorobenzoate, an intermediate in the metabolism of 4-chlorobiphenyl by Pseudomonas cepacia P166. Appl Environ Microbiol 61:443–447
Armengaud J, Happe B, Timmis KN (1998) Genetic analysis of dioxin dioxygenase of Sphingomonas sp. strain RW1: catabolic genes dispersed on the genome. J Bacteriol 180:3954–3966
Arnett CM, Parales JV, Haddock JD (2000) Influence of chlorine substituents on rates of oxidation of chlorinated biphenyls by the biphenyl dioxygenase of Burkholderia sp strain LB400. Appl Environ Microbiol 66:2928–2933
Asturias JA, Timmis KN (1993) Three different 2,3-dihydroxybiphenyl-1,2-dioxygenase genes in the gram-positive polychlorobiphenyl-degrading bacterium Rhodococcus globerulus P6. J Bacteriol 175:4631–4640
ATSDR (2000) Toxicological profile for polychlorinated biphenyls (PCBs). Agency for Toxic Substances and Disease Registry, United States Department of Health and Human Services, Public Health Service, Atlanta, GA
Babbitt PC, Kenyon GL, Martin BM, Charest H, Sylvestre M, Scholten JD, Chang K-H, Liang P-H, Dunaway-Mariano D (1992) Ancestry of the 4-chlorobenzoate dehalogenase: analysis of amino acid sequence identities among families of acyl: adenyl ligases, enoyl-CoA hydratases/isomerases, and acyl-CoA thioesterases. Biochemistry 31:5594–5604
Barriault D, Durand J, Maaroufi H, Eltis LD, Sylvestre M (1998) Degradation of polychlorinated biphenyl metabolites by naphthalene-catabolizing enzymes. Appl Environ Microbiol 64:4637–4642
Barriault D, Plante MM, Sylvestre M (2002) Family shuffling of a targeted bphA region to engineer biphenyl dioxygenase. J Bacteriol 184:3794–3800
Barriault D, Sylvestre M (1999a) Catalytic activity of Pseudomonas putida strain G7 naphthalene 1,2-dioxygenase on biphenyl. Int Biodeterior Biodegrad 44:33–37
Barriault D, Sylvestre M (1999b) Functionality of biphenyl 2,3-dioxygenase components in naphthalene 1,2-dioxygenase. Appl Microbiol Biotechnol 51:592–597
Barriault D, Vedadi M, Powlowski J, Sylvestre M (1999) cis-2,3-Dihydro-2,3-dihydroxybiphenyl dehydrogenase and cis-1,2-dihydro-1,2-dihydroxynaphathalene dehydrogenase catalyze dehydrogenation of the same range of substrates. Biochem Biophys Res Commun 260:181–187
Bartels F, Backhaus S, Moore ERB, Timmis KN, Hofer B (1999) Occurrence and expression of glutathione-S-transferase-encoding bphK genes in Burkholderia sp. strain LB400 and other biphenyl-utilizing bacteria. Microbiology 145:2821–2834
Bartels I, Knackmuss H-J, Reineke W (1984) Suicide inactivation of catechol 2,3-dioxygenase from Pseudomonas putida mt-2 by 3-halocatechols. Appl Environ Microbiol 47:500–505
Bayly RC, Dagley S, Gibson DT (1966) The metabolism of cresols by species of Pseudomonas. Biochem J 101:293–301
Bedard DL, Haberl ML (1990) Influence of chlorine substitution pattern on the degradation of polychlorinated biphenyls by eight bacterial strains. Microb Ecol 20:87–102
Bedard DL, Haberl ML, May RJ, Brennan MJ (1987) Evidence for novel mechanisms of polychlorinated biphenyl metabolism in Alcaligenes eutrophus H850. Appl Environ Microbiol 53:1103–1112
Beltrametti F, Reniero D, Backhaus S, Hofer B (2001) Analysis of transcription of the bph locus of Burkholderia sp strain LB400 and evidence that the ORF0 gene product acts as a regulator of the promoter. Microbiology 147:2169–2182
Benning MM, Wesenberg G, Liu RQ, Taylor KL, Dunaway-Mariano D, Holden HM (1998) The three-dimensional structure of 4-hydroxybenzoyl-CoA thioesterase from Pseudomonas sp. strain CBS-3. J Biol Chem 273:33572–33579
Billingsley KA, Backus SM, Ward OP (1999) Effect of surfactant solubilization on biodegradation of polychlorinated bipbenyl congeners by Pseudomonas LB400. Appl Microbiol Biotechnol 52:255–260
Blasco R, Mallavarapu M, Wittich RM, Timmis KN, Pieper DH (1997) Evidence that formation of protoanemonin from metabolites of 4-chlorobiphenyl degradation negatively affects the survival of 4-chlorobiphenyl-cometabolizing microorganisms. Appl Environ Microbiol 63:427–434
Blasco R, Wittich R-M, Mallavarapu M, Timmis KN, Pieper DH (1995) From xenobiotic to antibiotic. Formation of protoanemonin from 4-chlorocatechol by enzymes of the 3-oxoadipate pathway. J Biol Chem 270:29229–29235
Blumenroth P, Wagner-Döbler I (1998) Survival of inoculants in polluted sediments: effect of strain origin and carbon source competition. Microb Ecol 35:279–288
Brazil GM, Kenefick L, Callanan M, Haro A, Lorenzo V, Dowling DN, O’Gara F (1995) Construction of a rhizosphere pseudomonad with potential to degrade polychlorinated biphenyls and detection of bph gene expression in the rhizosphere. Appl Environ Microbiol 61:1946–1952
Broderick JB, O’Halloran TV (1991) Overproduction, purification, and characterization of chlorocatechol dioxygenase, a non-heme iron dioxygenase with broad substrate tolerance. Biochemistry 30:7349–7358
Brühlmann F, Chen W (1999a) Transformation of polychlorinated biphenyls by a novel BphA variant through the meta-cleavage pathway. FEMS Microbiol Lett 179:203–208
Brühlmann F, Chen W (1999b) Tuning biphenyl dioxygenase for extended substrate specificity. Biotechnol Bioeng 63:544–551
Burrus V, Pavlovic G, Decaris B, Guedon G (2002) Conjugative transposons: the tip of the iceberg. Mol Microbiol 46:601–610
Cámara B, Herrera C, González M, Couve E, Hofer B, Seeger M (2004) From PCBs to highly toxic metabolites by the biphenyl pathway. Environ Microbiol 6:842–850
Catelani D, Mosselmans G, Nienhaus J, Sorlini C, Treccani V (1970) Microbial degradation of aromatic hydrocarbons used as reactor coolants. Experientia 26:922–923
Chang KH, Liang PH, Beck W, Scholten JD, Dunaway-Mariano D (1992) Isolation and characterization of the three polypeptide components of 4-chlorobenzoate dehalogenase from Pseudomonas sp. strain CBS-3. Biochemistry 31:5605–5610
Chatterjee DK, Chakrabarty AM (1983) Genetic homology between independently isolated chlorobenzoate-degradative plasmids. J Bacteriol 153:532–534
Chatterjee DK, Kellogg ST, Hamada S, Chakrabarty AM (1981a) Plasmid specifying total degradation of 3-chlorobenzoate by a modified ortho pathway. J Bacteriol 146:639–646
Chatterjee DK, Kellogg ST, Watkins DR, Chakrabarty AM (1981b) Plasmids in the biodegradation of chlorinated aromatic compounds. In: Levy SB, Clowes RC, Koenig EL (eds) Molecular biology, pathogenicity, and ecology of bacterial plasmids. Plenum, New York, pp 519–528
Chun HK, Ohnishi Y, Shindo K, Misawa N, Furukawa K, Horinouchi S (2003) Biotransformation of flavone and flavanone by Streptomyces lividans cells carrying shuffled biphenyl dioxygenase genes. J Mol Catal B 21:113–121
Coco WM, Rothmel RK, Henikoff S, Chakrabarty AM (1993) Nucleotide sequence and initial functional characterization of the clcR gene encoding a LysR family activator of the clcABD chlorocatechol operon in Pseudomonas putida. J Bacteriol 175:417–427
Colores GM, Macur RE, Ward DM, Inskeep WP (2000) Molecular analysis of surfactant-driven microbial population shifts in hydrocarbon-contaminated soil. Appl Environ Microbiol 66:2959–2964
Cowles CE, Nichols NN, Harwood CS (2000) BenR, a XylS homologue, regulates three different pathways of aromatic acid degradation in Pseudomonas putida. J Bacteriol 182:6339–6346
Crooks GP, Xu L, Barkley RM, Copley SD (1995) Exploration of possible mechanisms for 4-chlorobenzoyl CoA dehalogenase: evidence for an aryl-enzyme intermediate. J Am Chem Soc 117:10791–10798
Dai S, Vaillancourt F, Maaroufi H, Drouin N, Neau D, Snieckus V, Bolin J, Eltis L (2002) Identification and analysis of a bottleneck in PCB biodegradation. Nat Struct Biol 9:934–939
Don RH, Pemberton JM (1981) Properties of six pesticide degradation plasmids isolated from Alcaligenes paradoxus and Alcaligenes eutrophus. J Bacteriol 145:681–686
Don RH, Pemberton JM (1985) Genetic and physical map of the 2,4-dichlorophenoxyacetic acid-degradative plasmid pJP4. J Bacteriol 161:466–468
Dong J, Carey PR, Wei YS, Luo LS, Lu XF, Liu RQ, Dunaway-Mariano D (2002) Raman evidence for Meisenheimer complex formation in the hydrolysis reactions of 4-fluorobenzoyl- and 4-nitrobenzoyl-coenzyme a catalyzed by 4-chlorobenzoyl-coenzyme A dehalogenase. Biochemistry 41:7453–7463
Donnelly PK, Hedge RS, Fletcher JS (1994) Growth of PCB-degrading bacteria on compounds from photosynthetic plants. Chemosphere 28:981–988
Dorn E, Knackmuss H-J (1978a) Chemical structure and biodegradability of halogenated aromatic compounds. Substituent effects on 1,2-dioxygenation of catechol. Biochem J 174:85–94
Dorn E, Knackmuss H-J (1978b) Chemical structure and biodegradability of halogenated aromatic compounds. Two catechol 1,2-dioxygenases from a 3-chlorobenzoate-grown pseudomonad. Biochem J 174:73–84
Drinker C, Warren M, Bennet G (1937) The problem of possible systemic effects from certain chlorinated hydrocarbons. J Ind Hyg Toxicol 19:283–311
Elsner A, Löffler F, Miyashita K, Müller R, Lingens F (1991) Resolution of 4-chlorobenzoate dehalogenase from Pseudomonas sp. strain CBS3 into three components. Appl Environ Microbiol 57:324–326
Eltis LD, Bolin JT (1996) Evolutionary relationships among extradiol dioxygenases. J Bacteriol 178:5930–5937
Engesser KH, Schulte P (1989) Degradation of 2-bromo-, 2-chloro- and 2-fluorobenzoate by Pseudomonas putida CLB 250. FEMS Microbiol Lett 60:143–148
Erickson BD, Mondello FJ (1992) Nucleotide sequencing and transcriptional mapping of the genes encoding biphenyl dioxygenase, a multicomponent polychlorinated-biphenyl-degrading enzyme in Pseudomonas strain LB400. J Bacteriol 174:2903–2912
Erickson BD, Mondello FJ (1993) Enhanced biodegradation of polychlorinated biphenyls after site-directed mutagenesis of a biphenyl dioxygenase gene. Appl Environ Microbiol 59:3858–3862
Eulberg D, Kourbatova EM, Golovleva LA, Schlömann M (1998) Evolutionary relationship between chlorocatechol catabolic enzymes from Rhodococcus opacus 1CP and their counterparts in proteobacteria: sequence divergence and functional convergence. J Bacteriol 180:1082–1094
Faroon O, Jones D, de Rosa C (2001) Effects of polychlorinated biphenyls on the nervous system. Toxicol Ind Health 16:305–333
Faroon O, Keith L, Smith-Simon C, De Rosa C (2003) Polychlorinated biphenyls. Human health aspects. In: Concise international chemical assessment document 55. World Health Organization, Geneva
Fava F, Piccolo A (2002) Effects of humic substances on the bioavailability and aerobic biodegradation of polychlorinated biphenyls in a model soil. Biotechnol Bioeng 77:204–211
Ferrer M, Golyshin P, Timmis KN (2003) Novel maltotriose esters enhance biodegradation of Aroclor 1242 by Burkholderia cepacia LB400. World J Microbiol Biotechnol 19:637–643
Fetzner S, Müller R, Lingens F (1989) Degradation of 2-chlorobenzoate by Pseudomonas cepacia 2CBS. Biol Chem Hoppe-Seyler 370:1173–1182
Fetzner S, Müller R, Lingens F (1992) Purification and some properties of 2-halobenzoate 1,2-dioxygenase, a two component enzyme system from Pseudomonas cepacia 2CBS. J Bacteriol 174:279–290
Focht D (1995) Strategies for the improvement of aerobic metabolism of polychlorinated biphenyls. Curr Opin Biotechnol 6:341–346
Fuenmayor SL, Wild M, Boyes AL, Williams PA (1998) A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J Bacteriol 180:2522–2530
Furukawa K, Hirose J, Suyama A, Zaiki T, Hayashida S (1993) Gene components responsible for discrete substrate specifity in the metabolism of biphenyl (bph operon) and toluene (tod operon). J Bacteriol 175:5224–5232
Furukawa K, Miyazaki T (1986) Cloning of a gene cluster encoding biphenyl and chlorobiphenyl degradation in Pseudomonas pseudoalcaligenes. J Bacteriol 166:392–398
Furukawa K, Simon JR, Chakrabarty AM (1983) Common induction and regulation of biphenyl, xylene/toluzene,m and salicylate catabolism catabolism in Pseudomonas paucimobilis. J Bacteriol 154:1356–1362
Furukawa K, Tomizuka N, Kamibayashi A (1979) Effect of chlorine substitution on the bacterial metabolism of various polychlorinated biphenyls. Appl Environ Microbiol 38:301–310
Gibson DT, Parales RE (2000) Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr Opin Biotechnol 11:236–243
Gibson DT, Roberts RL, Wells MC, Kobal VM (1973) Oxidation of biphenyl by a Beijerinckia species. Biochem Biophys Res Commun 50:211–215
Gilbert ES, Crowley DE (1997) Plant compounds that induce polychlorinated biphenyl biodegradation by Arthrobacter sp. strain B1B. Appl Environ Microbiol 63:1933–1938
Göbel M, Kranz OH, Kaschabek SR, Schmidt E, Pieper DH, Reineke W (2004) Microorganisms degrading chlorobenzene via a meta-cleavage pathway harbor highly similar chlorocatechol 2,3-dioxygenase-encoding gene clusters. Arch Microbiol 182:147–156
Golyshin PM, Fredrickson HL, Giuliano L, Rothmel R, Timmis KN, Yakimov MM (1999) Effect of novel biosurfactants on biodegradation of polychlorinated biphenyls by pure and mixed bacterial cultures. Microbiologica 22:257–267
Haak B, Fetzner S, Lingens F (1995) Cloning, nucleotide sequence, and expression of the plasmid-encoded genes for the two-component 2-halobenzoate 1,2-dioxygenase from Pseudomonas cepacia 2CBS. J Bacteriol 177:667–675
Haddock JD, Gibson DT (1995) Purification and characterization of the oxygenase component of biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J Bacteriol 177:5834–5839
Haddock JD, Horton JR, Gibson DT (1995) Dihydroxylation and dechlorination of chlorinated biphenyls by purified biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J Bacteriol 177:20–26
Harayama S, Rekik M, Bairoch A, Neidle EL, Ornston LN (1991) Potential DNA slippage structures acquired during evolutionary divergence of Acinetobacter calcoaceticus chromosomal benABC and Pseudomonas putida TOL pWW0 plasmid xylXYZ, genes encoding benzoate dioxygenases. J Bacteriol 173:7540–7548
Harwood CS, Parales RE (1996) The beta-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol 50:553–590
Havel J, Reineke W (1991) Total degradation of various chlorobiphenyls by cocultures and in vivo constructed hybrid pseudomonads. FEMS Microbiol Lett 78:163–170
Havel J, Reineke W (1992) Degradation of Aroclor 1221 and survival of strains in soil microcosms. Appl Microbiol Biotechnol 38:129–134
Havel J, Reineke W (1993) Degradation of Aroclor 1221 in soil by a hybrid pseudomonad. FEMS Microbiol Lett 108:211–218
Hayase N, Taira K, Furukawa K (1990) Pseudomonas putida KF715 bphABCD operon encoding biphenyl and polychlorinated biphenyl degradation: cloning analysis, and expression in soil bacteria. J Bacteriol 172:1160–1164
Hein P, Powlowski J, Barriault D, Hurtubise Y, Ahmad D, Sylvestre M (1998) Biphenyl-associated meta-cleavage dioxygenases from Comamonas testosteroni B-356. Can J Microbiol 44:42–49
Heiss G, Stolz A, Kuhm AE, Müller C, Klein J, Altenbuchner J, Knackmuss H-J (1995) Characterization of a 2,3-dihydroxybiphenyl dioxygenase from the naphthalenesulfonate-degrading bacterium strain BN6. J Bacteriol 177:5865–5871
Hernandez BS, Higson FK, Kondrat R, Focht DD (1991) Metabolism of and inhibition by chlorobenzoates in Pseudomonas putida P111. Appl Environ Microbiol 57:3361–3366
Hernandez BS, Koh SC, Chial M, Focht DD (1997) Terpene-utilizing isolates and their relevance to enhanced biotransformation of polychlorinated biphenyls in soil. Biodegradation 8:153–158
Hickey WJ, Brenner V, Focht DD (1992) Mineralization of 2-chloro- and 2,5-dichlorobiphenyl by Pseudomonas sp. strain UCR2. FEMS Microbiol Lett 98:175–180
Hickey WJ, Focht DD (1990) Degradation of mono-, di-, and trihalogenated benzoic acids by Pseudomonas aeruginosa JB2. Appl Environ Microbiol 56:3842–3850
Hickey WJ, Sabat G (2001) Integration of matrix-assisted laser desorption ionization-time of flight mass spectrometry and molecular cloning for the identification and functional characterization of mobile ortho-halobenzoate oxygenase genes in Pseudomonas aeruginosa strain JB2. Appl Environ Microbiol 67:5648–5655
Hickey WJ, Sabat G, Yuroff AS, Arment AR, Perez-Lesher J (2001) Cloning, nucleotide sequencing, and functional analysis of a novel, mobile cluster of biodegradation genes from Pseudomonas aeruginosa strain JB2. Appl Environ Microbiol 67:4603–4609
Hickey WJ, Searles DB, Focht DD (1993) Enhanced mineralization of polychlorinated biphenyls in soil inoculated with chlorobenzoate-degrading bacteria. Appl Environ Microbiol 59:1194–1200
Higson FK, Focht DD (1990) Degradation of 2-bromobenzoic acid by a strain of Pseudomonas aeruginosa. Appl Environ Microbiol 56:1615–1619
Higson FK, Focht DD (1992) Utilization of 3-chloro-2-methylbenzoic acid by Pseudomonas cepacia MB2 through the meta fission pathway. Appl Environ Microbiol 58:2501–2504
Hofer B, Backhaus S, Timmis KN (1994) The biphenyl/polychlorinated biphenyl-degradation locus (bph) of Pseudomonas sp. LB400 encodes four additional metabolic enzymes. Gene 144:9–16
Hoffmann D, Kleinsteuber S, Müller RH, Babel W (2003) A transposon encoding the complete 2,4-dichlorophenoxyacetic acid degradation pathway in the alkalitolerant strain Delftia acidovorans P4a. Microbiology 149:2545–2556
Hollender J, Dott W, Hopp J (1994) Regulation of chloro- and ethylphenol degradation in Comamonas testosteroni JH5. Appl Environ Microbiol 60:2330–2338
Hollender J, Hopp J, Dott W (1997) Degradation of 4-chlorophenol via the meta cleavage pathway by Comamonas testosteroni JH5. Appl Environ Microbiol 63:4567–4572
Hrywna Y, Tsoi TV, Maltseva OV, Quensen JF, Tiedje JM (1999) Construction and characterization of two recombinant bacteria that grow on ortho- and para-substituted chlorobiphenyls. Appl Environ Microbiol 65:2163–2169
Hülsmeyer M, Hecht H, Niefind K, Hofer B, Eltis L, Timmis K, Schomburg D (1998) Crystal structure of cis-biphenyl-2,3-dihydrodiol-2,3-dehydrogenase from a PCB degrader at 2.0 A resolution. Protein Sci 7:1286–1293
Janke D, Fritsche W (1979) Dechlorierung von 4-chlorphenol nach extradioler Ringspaltung durch Pseudomonas putida. Z Allg Mikrobiol 19:139–141
Kasai Y, Shindo K, Harayama S, Misawa N (2003) Molecular characterization and substrate preference of a polycyclic aromatic hydrocarbon dioxygenase from Cycloclasticus sp. strain A5. Appl Environ Microbiol 69:6688–6697
Kaschabek SR, Kasberg T, Müller D, Mars AE, Janssen DB, Reineke W (1998) Degradation of chloroaromatics: purification and characterization of a novel type of chlorocatechol 2,3-dioxygenase of Pseudomonas putida GJ31. J Bacteriol 180:296–302
Kaschabek SR, Reineke W (1992) Maleylacetate reductase of Pseudomonas sp. strain B13: dechlorination of chloromaleylacetates, metabolites in the degradation of chloroaromatic compounds. Arch Microbiol 158:412–417
Kaulmann U, Kaschabek SR, Schlömann M (2001) Mechanism of chloride elimination from 3-chloro- and 2,4-dichloro-cis,cis-muconate: new insight obtained from analysis of muconate cycloisomerase variant CatB-K169A. J Bacteriol 183:4551–4561
Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S (1998) Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6:571–586
Kersten P, Chapman PJ, Dagley S (1985) Enzymatic release of halogens or methanol from some substituted protocatechuic acids. J Bacteriol 162:693–697
Kersten P, Dagley S, Whittaker J, Arciero D, Lipscomb J (1982) 2-Pyrone-4,6-dicarboxylic acid, a catabolite of gallic acids in Pseudomonas species. J Bacteriol 152:1154–1162
Kikuchi Y, Yasukochi Y, Nagata Y, Fukuda M, Takagi M (1994) Nucleotide sequence and functional analysis of the meta-cleavage pathway involved in biphenyl and polychlorinated biphenyl degradation in Pseudomonas sp. strain KKS102. J Bacteriol 176:4269–4276
Kim E, Zylstra GJ (1995) Molecular and biochemical characterization of two meta-cleavage dioxygenase involved in biphenyl and m-xylene degradation by Beijerinckia sp. strain B1. J Bacteriol 177:3095–3103
Kim E, Zylstra GJ (1999) Functional analysis of genes involved in biphenyl, naphthalene, phenanthrene, and m-xylene degradation by Sphingomonas yanoikuyae B1. J Ind Microbiol Biotechnol 23:294–302
Kim SJ, Chun J, Bae KS, Kim YC (2000) Polyphasic assignment of an aromatic-degrading Pseudomonas sp., strain DJ77, in the genus Sphingomonas as Sphingomonas chungbukensis sp nov. Int J Syst Evol Microbiol 50:1641–1647
Kim SY, Jung JY, Lim YH, Ahn JH, Kim SI, Hur HG (2003) cis-2′,3′-Dihydrodiol production on flavone B-ring by biphenyl dioxygenase from Pseudomonas pseudoalcaligenes KF707 expressed in Escherichia coli. Antonie Van Leeuwenhoek 84:261–268
Kimbara K, Hashimoto T, Fukuda M, Koana T, Takagi M, Oishi M, Yano K (1989) Cloning and sequencing of two tandem genes involved in degradation of 2,3-dihydroxybiphenyl to benzoic acid in the polychlorinated biphenyl-degrading soil bacterium Pseudomonas sp. strain KKS102. J Bacteriol 171:2740–2747
Kimura N, Nishi A, Goto M, Furukawa K (1997) Functional analyses of a variety of chimeric dioxygenases constructed from two biphenyl dioxygenases that are similar structurally but different functionally. J Bacteriol 179:3936–3943
Kitagawa W, Miyauchi K, Masai E, Fukuda M (2001a) Cloning and characterization of benzoate catabolic genes in the gram-positive polychlorinated biphenyl degrader Rhodococcus sp strain RHA1. J Bacteriol 183:6598–6606
Kitagawa W, Suzuki A, Hoaki T, Masai E, Fukuda M (2001b) Multiplicity of aromatic ring hydroxylation dioxygenase genes in a strong PCB degrader, Rhodococcus sp. strain RHA1 demonstrated by denaturing gradient gel electrophoresis. Biosci Biotechnol Biochem 65:1907–1911
Kitayama A, Achioku T, Yanagawa T, Kanou K, Kikuchi M, Ueda H, Suzuki E, Nishimura H, Nagamune T, Kawakami Y (1996) Cloning and characterization of extradiol aromatic ring-cleavage dioxygenases from Pseudomonas aeruginosa JI104. J Ferment Bioeng 82:217–223
Klages U, Lingens F (1979) Degradation of 4-chlorobenzoic acid by a Nocardia species. FEMS Microbiol Lett 6:201–203
Klages U, Lingens F (1980) Degradation of 4-chlorobenzoic acid by a Pseudomonas sp. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg Abt 1 C 1:215–223
Klemba M, Jakobs B, Wittich R, Pieper D (2000) Chromosomal integration of the tcb chlorocatechol degradation pathway genes as a means of expanding the growth substrate range of bacteria to include haloaromatics. J Bacteriol 182:3255–3261
Kosono S, Maeda M, Fuji F, Arai H, Kudo T (1997) Three of the seven bphC genes of Rhodococcus erythropolis TA421, isolated from a termite ecosystem, are located on an indigenous plasmid associated with biphenyl degradation. Appl Environ Microbiol 63:3282–3285
Kozlovsky SA, Zaitsev GM, Kunc F, Gabriel J, Boronin AM (1993) Degradation of 2-chlorobenzoic and 2,5-dichlorobenzoic acids in pure culture by Pseudomonas stutzeri. Folia Microbiol 38:371–375
Krooneman J, Moore ERB, van Velzen JCL, Prins RA, Forney LJ, Gottschal JC (1998) Competition for oxygen and 3-chlorobenzoate between two aerobic bacteria using different degradation pathways. FEMS Microbiol Ecol 26:171–179
Krooneman J, Wieringa EBA, Moore ERB, Gerritse J, Prins RA, Gottschal JC (1996) Isolation of Alcaligenes sp. strain L6 at low oxygen concentrations and degradation of 3-chlorobenzoate via a pathway not involving (chloro)catechols. Appl Environ Microbiol 62:2427–2434
Kumamaru T, Suenaga H, Mitsuoka M, Watanabe T, Furukawa K (1998) Enhanced degradation of polychlorinated biphenyls by directed evolution of biphenyl dioxygenase. Nat Biotechnol 16:663–666
Labbe D, Garnon J, Lau PCK (1997) Characterization of the genes encoding a receptor-like histidine kinase and a cognate response regulator from a biphenyl/polychlorobiphenyl-degrading bacterium, Rhodococcus sp. strain M5. J Bacteriol 179:2772–2776
Lee J, Min KR, Kim Y-C, Kim C-K, Lim J-Y, Yoon H, Min K-H, Lee K-S, Kim Y (1995) Cloning of salicylate hydroxylase gene and catechol 2,3-dioxygenase gene and sequencing of an intergenic sequence between the two genes of Pseudomonas putida KF715. Biochem Biophys Res Commun 211:382–388
Leesong M, Henderson B, Gillig J, Schwab J, Smith J (1996) Structure of a dehydratase-isomerase from the bacterial pathway for biosynthesis of unsaturated fatty acids: two catalytic activities in one active site. Structure 4:253–264
Liang P-H, Yang G, Dunaway-Mariano D (1993) Specificity of 4-chlorobenzoyl coenzyme A dehalogenase catalyzed dehalogenation of halogenated aromatics. Biochemistry 32:12245–12250
Liu R-Q, Liang P-H, Scholten J, Dunaway-Mariano D (1995) Transient state kinetic analysis of the chemical intermediates formed in the enzymatic dehalogenation of 4-chlorobenzoyl coenzyme A. J Am Chem Soc 117:5003–5004
Liu S, Ogawa N, Miyashita K (2001) The chlorocatechol degradative genes, tfdT-CDEF, of Burkholderia sp. strain NK8 are involved in chlorobenzoate degradation and induced by chlorobenzoates and chlorocatechols. Gene 268:207–214
Lloyd-Jones G, Ogden RC, Williams PA (1995) Inactivation of 2,3-dihydroxybiphenyl 1,2-dioxygenase from Pseudomonas sp. strain CB406 by 3,4-dihydroxybiphenyl (4-phenylcatechol). Biodegradation 6:11–17
Löffler F, Lingens F, Müller R (1995) Dehalogenation of 4-chlorobenzoate. Characterisation of 4-chlorobenzoyl-coenzyme A dehalogenase from Pseudomonas sp. CBS3. Biodegradation 6:203–212
Löffler F, Müller R (1991) Identification of 4-chlorobenzoyl-coenzyme A as intermediate in the dehalogenation catalyzed by 4-chlorobenzoate dehalogenase from Pseudomonas sp CBS3. FEBS Lett 290:224–226
Löffler F, Müller R, Lingens F (1992) Purification and properties of 4-halobenzoate-coenzyme A ligase from Pseudomonas sp. CBS3. Biol Chem Hoppe-Seyler 373:1001–1007
Lünsdorf H, Erb R, Abraham W, Timmis K (2000) ‘Clay hutches’: a novel interaction between bacteria and clay minerals. Environ Microbiol 2:161–168
Lunt D, Evans WC (1970) The microbial metabolism of biphenyl. Biochem J 118:54–55
Maeda M, Chung S-Y, Song E, Kudo T (1995) Multiple genes encoding 2,3-dihydroxybiphenyl 1,2-dioxygenase in the gram-positive polychlorinated biphenyl-degrading bacterium Rhodococcus erythropolis TA421, isolated from a termite ecosystem. Appl Environ Microbiol 61:549–555
Maeda T, Takahashi Y, Suenaga H, Suyama A, Goto M, Furukawa K (2001) Functional analyses of Bph-Tod hybrid dioxygenase, which exhibits high degradation activity toward trichloroethylene. J Biol Chem 276:29833–29838
Maltseva OV, Solyanikova IP, Golovleva LA (1994a) Chlorocatechol 1,2-dioxygenase from Rhodococcus erythropolis 1 CP. Kinetic and immunochemical comparison with analogous enzymes from Gram-negative strains. Eur J Biochem 226:1053–1061
Maltseva OV, Solyanikova IP, Golovleva LA, Schlömann M, Knackmuss H-J (1994b) Dienelactone hydrolase from Rhodococcus erythropolis 1 CP: purification and properties. Arch Microbiol 162:386–374
Mars AE, Kasberg T, Kaschabek SR, van Agteren MH, Janssen DB, Reineke W (1997) Microbial degradation of chloroaromatics: use of the meta-cleavage pathway for mineralization of chlorobenzene. J Bacteriol 179:4530–4537
Mars AE, Kingma J, Kaschabek SR, Reineke W, Janssen DB (1999) Conversion of 3-chlorocatechol by various catechol 2,3-dioxygenases and sequence analysis of the chlorocatechol dioxygenase region of Pseudomonas putida GJ31. J Bacteriol 181:1309–1318
Masai E, Sugiyama K, Iwashita N, Shimizu S, Hauschild JE, Hatta T, Kimbara K, Yano K, Fukuda M (1997) The bphDEF meta-cleavage pathway genes involved in biphenyl/polychlorinated biphenyl degradation are located on a linear plasmid and separated from the initial bphACB genes in Rhodococcus sp. strain RHA1. Gene 187:141–149
Masai E, Yamada A, Healy JM, Hatta T, Kimbara K, Fukuda M, Yano K (1995) Characterization of biphenyls catabolic genes of gram-positive polychlorinated biphenyls degrader Rhodococcus sp. strain RHA1. Appl Environ Microbiol 61:2079–2085
Mayes B, McConnell E, Neal B, Brunner M, Hamilton S, Sullivan T, Peters A, Ryan M, Toft J, Singer A, Brown J, Menton R, Moore J (1998) Comparative carcinogenicity in Sprague-Dawley rats of the polychlorinated biphenyl mixtures aroclors 1016, 1242, 1254, and 1260. Toxicol Sci 41:62–76
McCullar MV, Brenner V, Adams RH, Focht DD (1994) Construction of a novel polychlorinated biphenyl-degrading bacterium: utilization of 3,4-dichlorobiphenyl by Pseudomonas acidovorans M3GY. Appl Environ Microbiol 60:3833–3839
McFall SM, Parsek MR, Chakrabarty AM (1997) 2-Chloromuconate and ClcR-mediated activation of the clcABD operon: in vitro transcriptional and DNase I footprint analyses. J Bacteriol 179:3655–3663
McKay DB, Prucha M, Reineke W, Timmis KN, Pieper DH (2003) Substrate specificity and expression of three 2,3-dihydroxybiphenyl 1,2-dioxygenases from Rhodococcus globerulus strain P6. J Bacteriol 185:2944–2951
McKay DB, Seeger M, Zielinski M, Hofer B, Timmis KN (1997) Heterologous expression of biphenyl dioxygenase-encoding genes from a gram-positive broad-spectrum polychlorinated biphenyl degrader and characterization of chlorobiphenyl oxidation by the gene products. J Bacteriol 179:1924–1930
Meer JR van der, Ravatn R, Sentchilo V (2001) The clc element of Pseudomonas sp. strain B13 and other mobile degradative elements employing phage-like integrases. Arch Microbiol 175:79–85
Meer JR van der, van Neerven ARW, de Vries EJ, de Vos WM, Zehnder AJB (1991) Cloning and characterization of plasmid-encoded genes for the degradation of 1,2-dichloro-, 1,4-dichloro-, and 1,2,4-trichlorobenzene of Pseudomonas sp. strain P51. J Bacteriol 173:6–15
Merlin C, Springael D, Mergeay M, Toussaint A (1997) Organisation of the bph gene cluster of transposon Tn4371, encoding enzymes for the degradation of biphenyl and 4-chlorobiphenyl compounds. Mol Gen Genet 253:499–506
Merlin C, Springael D, Toussaint A (1999) Tn4371: a modular structure encoding a phage-like integrase, a Pseudomonas-like catabolic pathway, and RP4/Ti-like transfer functions. Plasmid 40:54
Moiseeva OV, Belova OV, Solyanikova IP, Schlömann M, Golovleva LA (2001) Enzymes of a new modified ortho-pathway utilizing 2-chlorophenol in Rhodococcus opacus 1CP. Biochemistry (Moscow) 66:548–555
Moiseeva OV, Linko EV, Baskunov BP, Golovleva LA (1999) Degradation of 2-chlorophenol and 3-chlorobenzoate by Rhodococcus opacus 1CP. Microbiology (Moscow) 68:400–405
Moiseeva OV, Solyanikova IP, Kaschabek SR, Groning J, Thiel M, Golovleva LA, Schlömann M (2002) A new modified ortho cleavage pathway of 3-chlorocatechol degradation by Rhodococcus opacus 1CP: genetic and biochemical evidence. J Bacteriol 184:5282–5292
Mokross H, Schmidt E, Reineke W (1990) Degradation of 3-chlorobiphenyl by in vivo constructed hybrid pseudomonads. FEMS Microbiol Lett 71:179–186
Mondello FJ (1989) Cloning and expression in Escherichia coli of Pseudomonas strain LB400 genes encoding polychlorinated biphenyl degradation. J Bacteriol 171:1725–1732
Mondello FJ, Turcich MP, Lobos JH, Erickson BD (1997) Identification and modification of biphenyl dioxygenase sequences that determine the specificity of polychlorinated biphenyl degradation. Appl Environ Microbiol 63:3096–3103
Mouz S, Merlin C, Springael D, Toussaint A (1999) A GntR-like negative regulator of the biphenyl degradation genes of the transposon Tn4371. Mol Gen Genet 262:790–799
Murray K, Duggleby CJ, Sala-Trepat JM, Williams PA (1972) The metabolism of benzoate and methylbenzoates via the meta-cleavage by Pseudomonas arvilla mt-2. Eur J Biochem 28:301–310
Nakatsu C, Wyndham RC (1993) Cloning and expression of the transposable chlorobenzoate-3,4-dioxygenase genes of Alcaligenes sp. BR60. Appl Environ Microbiol 59:3625–3633
Nakatsu CH, Providenti M, Wyndham RC (1997) The cis-diol dehydrogenase cbaC gene of Tn5271 is required for growth on 3-chlorobenzoate but not 3,4-dichlorobenzoate. Gene 196:209–218
Nakatsu CH, Straus NA, Wyndham RC (1995) The nucleotide sequence of the Tn5271 3-chlorobenzoate 3,4-dioxygenase genes (cbaAB) unites the class IA oxygenase in a single lineage. Microbiology 141:485–495
Narasimhan K, Basheer C, Bajic V, Swarup S (2003) Enhancement of plant-microbe interactions using a rhizosphere metabolomics-driven approach and its application in the removal of polychlorinated biphenyls. Plant Physiol 132:146–153
Nikodem P, Hecht V, Schlömann M, Pieper DH (2003) New bacterial pathway for 4- and 5-chlorosalicylate degradation via 4-chlorocatechol and maleylacetate in Pseudomonas sp. strain MT1. J Bacteriol 185:6790–6800
Nishi A, Tominaga K, Furukawa K (2000) A 90-kilobase conjugative chromosomal element coding for biphenyl and salicylate catabolism in Pseudomonas putida KF715. J Bacteriol 182:1949–1955
Ogawa N, McFall SM, Klem TJ, Miyashita K, Chakrabarty AM (1999) Transcriptional activation of the chlorocatechol degradative genes of Ralstonia eutropha NH9. J Bacteriol 181:6697–6705
Ogawa N, Miyashita K (1999) The chlorocatechol-catabolic transposon Tn5707 of Alcaligenes eutrophus NH9, carrying a gene cluster highly homologous to that in the 1,2,4-trichlorobenzene-degrading bacterium Pseudomonas sp. strain P51, confers the ability to grow on 3-chlorobenzoate. Appl Environ Microbiol 65:724–731
Ohta Y, Maeda M, Kudo T (2001) Pseudomonas putida CE2010 can degrade biphenyl by a mosaic pathway encoded by the tod operon and cmtE, which are identical to those of P. putida F1 except for a single base difference in the operator-promoter region of the cmt operon. Microbiology 147:31–41
Ohtsubo Y, Delawary M, Kimbara K, Takagi M, Ohta A, Nagata Y (2001) BphS, a key transcriptional regulator of bph genes involved in polychlorinated biphenyl/biphenyl degradation in Pseudomonas sp. KKS102. J Biol Chem 276:36146–36154
Parales RE, Lee K, Resnick SM, Jiang HY, Lessner DJ, Gibson DT (2000) Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme. J Bacteriol 182:1641–1649
Patel TR, Gibson DT (1974) Purification and properties of (+)-cis-naphthalene dihydrodiol dehydrogenase of Pseudomonas putida. J Bacteriol 119:879–888
Pavlu L, Vosahlova J, Klierova H, Prouza M, Demnerova K, Brenner V (1999) Characterization of chlorobenzoate degraders isolated from polychlorinated biphenyl-contaminated soil and sediment in the Czech Republic. J Appl Microbiol 87:381–386
Peloquin L, Greer CW (1993) Cloning and expression of the polychlorinated biphenyl-degradation gene cluster from Arthrobacter M5 and comparison to analogous genes from gram-negative bacteria. Gene 125:35–40
Pelz O, Tesar M, Wittich RM, Moore ERB, Timmis KN, Abraham WR (1999) Towards elucidation of microbial community metabolic pathways: unravelling the network of carbon sharing in a pollutant-degrading bacterial consortium by immunocapture and isotopic ratio mass spectrometry. Environ Microbiol 1:167–174
Perez-Pantoja D, Ledger T, Pieper DH, Gonzalez B (2003) Efficient turnover of chlorocatechols is essential for growth of Ralstonia eutropha JMP134(pJP4) in 3-chlorobenzoic acid. J Bacteriol 185:1534–1542
Pieper DH, Knackmuss H-J, Timmis KN (1993) Accumulation of 2-chloromuconate during metabolism of 3-chlorobenzoate by Alcaligenes eutrophus JMP134. Appl Microbiol Biotechnol 39:563–567
Pinyakong O, Habe H, Omori T (2003) The unique aromatic catabolic genes in sphingomonads degrading polycyclic aromatic hydrocarbons (PAHs). J Gen Appl Microbiol 49:1–19
Poh RPC, Smith ARW, Bruce IJ (2002) Complete characterisation of Tn5530 from Burkholderia cepacia strain 2a (pIJB1) and studies of 2,4-dichlorophenoxyacetate uptake by the organism. Plasmid 48:1–12
Potrawfke T, Armengaud J, Wittich RM (2001) Chlorocatechols at positions 4 and 5 are substrates of the broad-spectrum chlorocatechol 1,2-dioxygenase Pseudomonas chlororaphis RW71. J Bacteriol 183:997–1011
Providenti MA, Wyndham RC (2001) Identification and functional characterization of CbaR, a MarR-like modulator of the cbaABC-encoded chlorobenzoate catabolism pathway. Appl Environ Microbiol 67:3530–3541
Prucha M, Peterseim A, Timmis KN, Pieper DH (1996a) Muconolactone isomerase of the 3-oxoadipate pathway catalyzes dechlorination of 5-chlorosubstituted muconolactones. Eur J Biochem 237:350–356
Prucha M, Wray V, Pieper DH (1996b) Metabolism of 5-chlorosubstituted muconolactones. Eur J Biochem 237:357–366
Raschke H, Fleischmann T, van der Meer JR, Kohler HPE (1999) cis-Chlorobenzene dihydrodiol dehydrogenase (TcbB) from Pseudomonas sp strain P51, expressed in Escherichia coli DH5 alpha(PTCB149), catalyzes enantioselective dehydrogenase reactions. Appl Environ Microbiol 65:5242–5246
Raschke H, Meier M, Burken JG, Hany R, Muller MD, Van der Meer JR, Kohler HPE (2001) Biotransformation of various substituted aromatic compounds to chiral dihydrodihydroxy derivatives. Appl Environ Microbiol 67:3333–3339
Ravatn R, Studer S, Springael D, Zehnder AJB, van der Meer JR (1998a) Chromosomal integration, tandem amplification, and deamplification in Pseudomonas putida F1 of a 105-kilobase genetic element containing the chlorocatechol degradative genes from Pseudomonas sp. strain B13. J Bacteriol 180:4360–4369
Ravatn R, Studer S, Zehnder AJB, van der Meer JR (1998b) Int-B13, an unusual site-specific recombinase of the bacteriophage P4 integrase family, is responsible for chromosomal insertion of the 105-kilobase clc element of Pseudomonas sp. strain B13. J Bacteriol 180:5505–5514
Ravatn R, Zehnder AJB, van der Meer JR (1998c) Low-frequency horizontal transfer of an element containing the chlorocatechol degradation genes from Pseudomonas sp. strain B13 to Pseudomonas putida F1 and to indigenous bacteria in laboratory-scale activated-sludge microcosms. Appl Environ Microbiol 64:2126–2132
Reineke W (1998) Development of hybrid strains for the mineralization of chloroaromatics by patchwork assembly. Annu Rev Microbiol 52:287–331
Reineke W, Knackmuss H-J (1978a) Chemical structure and biodegradability of halogenated aromatic compounds. Substituent effects on 1,2-dioxygenation of benzoic acid. Biochim Biophy Acta 532:412–423
Reineke W, Knackmuss H-J (1978b) Chemical structure and biodegradability of halogenated aromatic compounds. Substituent effects on dehydrogenation of 3,5-cyclohexadiene-1,2-diol-1-carboxylic acid. Biochim Biophys Acta 542:424–429
Reineke W, Knackmuss H-J (1979) Construction of haloaromatics utilising bacteria. Nature 277:385–386
Rodrigues J, Maltseva O, Tsoi T, Helton R, Quensen J, Fukuda M, Tiedje J (2001) Development of a Rhodococcus recombinant strain for degradation of products from anaerobic dechlorination of PCBs. Environ Sci Technol 35:663–668
Rogers JE, Gibson DT (1977) Purification and properties of cis-toluene dihydrodiol dehydrogenase from Pseudomonas putida. J Bacteriol 130:1117–1124
Romanov V, Hausinger RP (1994) Pseudomonas aeruginosa 142 uses a three-component ortho-halobenzoate 1,2-dioxygenase for the metabolism of 2,4-dichloro- and 2-chlorobenzoate. J Bacteriol 176:3368–3374
Romanov VP, Grechkina GM, Adanin VM, Starovoitov II (1993) Oxidative dehalogenation of 2-chloro- and 2,4-dichlorobenzoates by Pseudomonas aeruginosa. Microbiology 62:532–536
Romine MF, Stillwell LC, Wong KK, Thurston SJ, Sisk EC, Sensen C, Gaasterland T, Fredrickson JK, Saffer JD (1999) Complete sequence of a 184-kilobase catabolic plasmid from Sphingomonas aromaticivorans F199. J Bacteriol 181:1585–1602
Ron EZ, Rosenberg E (2001) Natural roles of biosurfactants. Environ Microbiol 3:229–236
Ruisinger S, Klages U, Lingens F (1976) Abbau der 4-Chlorbenzoesäure durch eine Arthrobacter-Species. Arch Microbiol 110:253–256
Sakai M, Masai E, Asami H, Sugiyama K, Kimbara K, Fukuda M (2002) Diversity of 2,3-dihydroxybiphenyl dioxygenase genes in a strong PCB degrader, Rhodococcus sp. strain RHA1. J Biosci Bioeng 93:421–427
Schell U, Helin S, Kajander T, Schlömann M, Goldman A (1999) Structural basis for the activity of two muconate cycloisomerase variants toward substituted muconates. Proteins 34:125–136
Schlömann M (1994) Evolution of chlorocatechol catabolic pathways. Biodegradation 5:301–321
Schlömann M, Schmidt E, Knackmuss H-J (1990) Different types of dienelactone hydrolase in 4-fluorobenzoate-utilizing bacteria. J Bacteriol 172:5112–5118
Schmidt E, Knackmuss H-J (1980) Chemical structure and biodegradability of halogenated aromatic compounds. Conversion of chlorinated muconic acids into maleoylacetic acid. Biochem J 192:339–347
Schmidt E, Remberg G, Knackmuss H-J (1980) Chemical structure and biodegradability of halogenated aromatic compounds. Halogenated muconic acids as intermediates. Biochem J 192:331–337
Schweigert N, Zehnder AJB, Eggen RIL (2001) Chemical properties of catechols and their molecular modes of toxic action in cells, from microorganisms to mammals. Environ Microbiol 3:81–91
Seah SYK, Labbe G, Nerdinger S, Johnson MR, Snieckus V, Eltis LD (2000) Identification of a serine hydrolase as a key determinant in the microbial degradation of polychlorinated biphenyls. J Biol Chem 275:15701–15708
Seegal B, Holden M (1945) The antibiotic activity of extracts of Ranunculaceae. Science 101:413–414
Seeger M, Camara B, Hofer B (2001) Dehalogenation, denitration, dehydroxylation, and angular attack on substituted biphenyls and related compounds by a biphenyl dioxygenase. J Bacteriol 183:3548–3555
Seeger M, Gonzalez M, Camara B, Munoz L, Ponce E, Mejias L, Mascayano C, Vasquez Y, Sepulveda-Boza S (2003) Biotransformation of natural and synthetic isoflavonoids by two recombinant microbial enzymes. Appl Environ Microbiol 69:5045–5050
Seeger M, Timmis KN, Hofer B (1995) Degradation of chlorobiphenyls catalyzed by the bph-encoded biphenyl-2,3-dioxygenase and biphenyl-2,3-dihydrodiol-2,3-dehydrogenase of Pseudomonas sp. LB400. FEMS Microbiol Lett 133:259–264
Seeger M, Timmis KN, Hofer B (1997) Bacterial pathways for the degradation of polychlorinated biphenyls. Marine Chem 58:327–333
Seeger M, Zielinski M, Timmis KN, Hofer B (1999) Regiospecificity of dioxygenation of di- to pentachlorobiphenyls and their degradation to chlorobenzoates by the bph-encoded catabolic pathway of Burkholderia sp. strain LB400. Appl Environ Microbiol 65:3614–3621
Sentchilo V, Ravatn R, Werlen C, Zehnder AJB, van der Meer JR (2003) Unusual integrase gene expression on the clc genomic island in Pseudomonas sp strain B13. J Bacteriol 185:4530–4538
Shimizu S, Kobayashi H, Masai E, Fukuda M (2001) Characterization of the 450-kb linear plasmid in a polychlorinated biphenyl degrader, Rhodococcus sp. strain RHA1. Appl Environ Microbiol 67:2021–2028
Shindo K, Ohnishi Y, Chun HK, Takahashi H, Hayashi M, Saito A, Iguchi K, Furukawa K, Harayama S, Horinouchi S, Misawa N (2001) Oxygenation reactions of various tricyclic fused aromatic compounds using Escherichia coli and Streptomyces lividans transformants carrying several arene dioxygenase genes. Biosci Biotechnol Biochem 65:2472–2481
Shingler V, Powlowski J, Marklund U (1992) Nucleotide sequence and functional analysis of the complete phenol/3,4-dimethylphenol catabolic pathway of Pseudomonas sp. strain CF600. J Bacteriol 174:711–724
Skiba A, Hecht V, Pieper DH (2002) Formation of protoanemonin from 2-chloro-cis,cis-muconate by the combined action of muconate cycloisomerase and muconolactone isomerase. J Bacteriol 184:5402–5409
Solyanikova IP, Malteva OV, Vollmer MD, Golovleva LA, Schlömann M (1995) Characterization of muconate and chloromuconate cycloisomerase from Rhodococcus erythropolis 1CP: indications for functionally convergent evolution among bacterial cycloisomerases. J Bacteriol 177:2821–2826
Sondossi M, Sylvestre M, Ahmad D (1992) Effects of chlorobenzoate transformation on the Pseudomonas testosteroni biphenyl and chlorobiphenyl degradation pathway. Appl Environ Microbiol 58:485–495
Springael D, Kreps S, Mergeay M (1993) Identification of a catabolic transposon, Tn4371, carrying biphenyl and 4-chlorobiphenyl degradation genes in Alcaligenes eutrophus A5. J Bacteriol 175:1674–1681
Springael D, Peys K, Ryngaert A, Van Roy S, Hooyberghs L, Ravatn R, Heyndrickx M, van der Meer JR, Vandecasteele C, Mergeay M, Diels L (2002) Community shifts in a seeded 3-chlorobenzoate degrading membrane biofilm reactor: indications for involvement of in situ horizontal transfer of the clc-element from inoculum to contaminant bacteria. Environ Microbiol 4:70–80
Stanier RY, Ornston LN (1973) The β-ketoadipate pathway. In: Advances in microbial physiology. Academic, London, pp 89–151
Stecker C, Johann A, Herzberg C, Averhoff B, Gottschalk G (2003) Complete nucleotide sequence and genetic organization of the 210-kilobase linear plasmid of Rhodococcus erythropolis BD2. J Bacteriol 185:5269–5274
Stelmack PL, Gray MR, Pickard MA (1999) Bacterial adhesion to soil contaminants in the presence of surfactants. Appl Environ Microbiol 65:163–168
Stratford J, Wright M, Reineke W, Mokross H, Havel J, Knowles C, Robinson G (1996) Influence of chlorobenzoates on the utilization of chlorobiphenyls and chlorobenzoate mixtures by chlorobiphenyl/chlorobenzoate-mineralising hybrid bacterial strains. Arch Microbiol 165:213–218
Suenaga H, Goto M, Furukawa K (2001a) Emergence of multifunctional oxygenase activities by random priming recombination. J Biol Chem 276:22500–22506
Suenaga H, Mitsuoka M, Ura Y, Watanabe T, Furukawa K (2001b) Directed evolution of biphenyl dioxygenase: emergence of enhanced degradation capacity for benzene, toluene, and alkylbenzenes. J Bacteriol 183:5441–5444
Suenaga H, Nishi A, Watanabe T, Sakai M, Furukawa K (1999) Engineering a hybrid pseudomonad to acquire 3,4-dioxygenase activity for polychlorinated biphenyls. J Biosci Bioeng 87:430–435
Suenaga H, Watanabe T, Sato M, Ngadiman, Furukawa K (2002) Alteration of regiospecificity in biphenyl dioxygenase by active-site engineering. J Bacteriol 184:3682–3688
Suzuki K, Ogawa N, Miyashita K (2001) Expression of 1,2-halobenzoate dioxygenase genes (cbdSABC) involved in the degradation of benzoate and 2-halobenzoate in Burkholderia sp. TH2. Gene 262:137–145
Taguchi K, Motoyama M, Kudo T (2004) Multiplicity of 2,3-dihydroxybiphenyl dioxygenase genes in the Gram-positive polychlorinated biphenyl degrading bacterium Rhodococcus rhodochrous K37. Biosci Biotechnol Biochem 68:787–795
Taira K, Hayase N, Arimura N, Yamashita S, Miyazaki T, Furukawa K (1988) Cloning and nucleotide sequence of the 2,3-dihydroxybiphenyl dioxygenase gene from the PCB-degrading strain of Pseudomonas paucimobilis Q1. Biochemistry 27:3990–3996
Takeda H, Yamada A, Miyauchi K, Masai E, Fukuda M (2004) Characterization of transcriptional regulatory genes for biphenyl degradation in Rhodococcus sp. strain RHA1. J Bacteriol 186:2134–2146
Tandlich R, Brezna B, Dercova K (2001) The effect of terpenes on the biodegradation of polychlorinated biphenyls by Pseudomonas stutzeri. Chemosphere 44:1547–1555
Toussaint A, Merlin C, Monchy S, Benotmane MA, Leplae R, Mergeay M, Springael D (2003) The biphenyl- and 4-chlorobiphenyl-catabolic transposon Tn4371, a member of a new family of genomic islands related to IncP and Ti plasmids. Appl Environ Microbiol 69:4837–4845
Trefault N, de la Iglesia R, Molina A, Manzano M, Ledger T, Perez-Pantoja D, Sanchez M, Stuardo M, Gonzalez B (2004) Genetic organization of the catabolic plasmid pJP4 from Ralstonia eutropha JMP134 (pJP4) reveals mechanisms of adaptation to chloroaromatic pollutants and evolution of specialized chloroaromatic degradation pathways. Environ Microbiol 6:655–668
Triska J, Kuncova G, Mackova M, Novakova H, Paasivirta J, Lahtipera M, Vrchotova N (2004) Isolation and identification of intermediates from biodegradation of low chlorinated biphenyls (Delor-103). Chemosphere 54:725–733
Tsoi TV, Plotnikova EG, Cole JR, Guerin WF, Bagdasarian M, Tiedje JM (1999) Cloning, expression, and nucleotide sequence of the Pseudomonas aeruginosa 142 ohb genes coding for oxygenolytic ortho dehalogenation of halobenzoates. Appl Environ Microbiol 65:2151–2162
Vaillancourt FH, Labbe G, Drouin NM, Fortin PD, Eltis LD (2002) The mechanism-based inactivation of 2,3-dihydroxybiphenyl 1,2-dioxygenase by catecholic substrates. J Biol Chem 277:2019–2027
Vaillancourt FH, Haro M, Drouin N, Karim Z, Maaroufi H, Eltis L (2003) Characterization of extradiol dioxygenases from a polychlorinated biphenyl-degrading strain that possess higher specificities for chlorinated metabolites. J Bacteriol 185:1253–1260
Vedler E, Koiv V, Heinaru A (2000) Analysis of the 2,4-dichlorophenoxyacetic acid-degradative plasmid pEST4011 of Achromobacter xylosooxidans subsp denitrificans strain EST4002. Gene 255:281–288
Vollmer MD, Schlömann M (1995) Conversion of 2-chloro-cis,cis-muconate and its metabolites 2-chloro- and 5-chloromuconolactone by chloromuconate cycloisomerase of pJP4 and pAC27. J Bacteriol 177:2938–2941
Vollmer MD, Fischer P, Knackmuss H-J, Schlömann M (1994) Inability of muconate cycloisomerases to cause dehalogenation during conversion of 2-chloro-cis,cis-muconate. J Bacteriol 176:4366–4375
Watanabe T, Fujihara H, Furukawa K (2003) Characterization of the second LysR-type regulator in the biphenyl-catabolic gene cluster of Pseudomonas pseudoalcaligenes KF707. J Bacteriol 185:3575–3582
Watanabe T, Inoue R, Kimura N, Furukawa K (2000) Versatile transcription of biphenyl catabolic bph operon in Pseudomonas pseudoalcaligenes KF707. J Biol Chem 275:31016–31023
Worsey M, Williams PA (1975) Metabolism of toluene and xylenes by Pseudomonas putida (arvilla) mt-2: evidence for a new function of the TOL plasmid. J Bacteriol 124:7–13
Yamada A, Kishi H, Sugiyama K, Hatta T, Nakamura K, Masai E, Fukuda M (1998) Two nearly identical aromatic compound hydrolase genes in a strong polychlorinated biphenyl degrader, Rhodococcus sp. strain RHA1. Appl Environ Microbiol 64:2006–2012
Yang G, Liang P-H, Dunaway-Mariano D (1994) Evidence of nucleophilic catalysis in the aromatic substitution reaction catalyzed by (4-chlorobenzoyl) coenzyme A dehalogenase. Biochemistry 33:8527–8531
Zaitsev GM, Karasevich YN (1984) Utilization of 2-chlorobenzoic acid by Pseudomonas cepacia. Mikrobiologiya 53:75–80
Zielinski M, Backhaus S, Hofer B (2002) The principal determinants for the structure of the substrate-binding pocket are located within a central core of a biphenyl dioxygenase alpha subunit. Microbiology 148:2439–2448
Zielinski M, Kahl S, Hecht HJ, Hofer B (2003) Pinpointing biphenyl dioxygenase residues that are crucial for substrate interaction. J Bacteriol 185:6976–6980
Zylstra GJ, Kim E (1997) Aromatic hydrocarbon degradation by Sphingomonas yanoikuyae B1. J Ind Microbiol Biotechnol 19:408–414
Zylstra GJ, McCombie WR, Gibson DT, Finette BA (1988) Toluene degradation by Pseudomonas putida F1: genetic organization of the tod operon. Appl Environ Microbiol 54:1498–1503
Acknowledgements
The work of D.H.P. in biodegradation is supported by grants EVK1-CT-1999-00023, QLK3-CT-2000-00731 and ICA4-CT-2002-10011 from the EU and by the DFG-European Graduate College 653 contract. Critical reading of the manuscript by Lotte Gabriel-Jürgens and Peter Golyshin is gratefully acknowledged
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pieper, D.H. Aerobic degradation of polychlorinated biphenyls. Appl Microbiol Biotechnol 67, 170–191 (2005). https://doi.org/10.1007/s00253-004-1810-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1810-4