
Abstract
The glyceraldehyde-3-phosphate dehydrogenase gene of Flammulina velutipes was isolated. The complete gpd sequence (from ATG to TAA) was 1,489 bp in length and contained nine introns. The locations of these nine introns were similar to those of other basidiomycetes, which might reflect the evolutionary divergence of these mushrooms. The F. velutipes gpd gene was found to encode a protein of 339 amino acids and its putative amino acid sequence revealed a high similarity to an analogous protein deriving from other basidiomycetes. Results of Southern blot analysis suggested that there existed only one copy of the gpd gene in the genome of F. velutipes and that there was one typical TATA box and two CAAT boxes located in the 5′ flanking region. The F. velutipes gpd promoter was fused to a hygromycin B phosphotransferase gene (hph) derived from Escherichia coli as a selection marker. Using the resulting construction, hph was efficiently transformed into F. velutipes by basidiospore electroporation. No false-positive antibiotic-resistant cultures were detected by PCR amplification and the hygromycin resistance trait was maintained stably during mitotic cell division for 3 months. Southern analysis of transformants indicated the integration of gene might occur by non-homologous recombination. This rapid and convenient electroporation procedure offers new prospects for the genetic manipulation and a tool for tagging genes of this important edible mushroom species. Sequence data will appear in the DDBJ/EMBL/GenBank nucleotide sequence database under accession number AF515622.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Using strong promoters to express heterologous genes in appropriate hosts is a major strategy in biotechnological applications. The glyceraldehyde-3-phosphate dehydrogenase (GPD, EC 1.2.1.12) promoter is deemed a promising candidate. GPD is one of the key enzymes in the glycolytic and gluconeogenesis pathways and comprises up to 5% of the soluble cellular protein content in Saccharomyces cerevisiae and other higher eukaryotes (Piechaczyk et al. 1984). Furthermore, gpd mRNA accounts for 2–5% of the poly (A)+ RNA present in yeasts (Holland and Holland 1978). These observations suggest that the gpd gene is regulated by a highly active promoter. In fact, vectors carrying the homologous gpd promoter region have been reported to be efficient in directing the expression of heterologous genes in yeasts (Bitter and Egan 1988; Doring et al. 1998; Eriksson et al. 1995; Vassileva et al. 2001) and filamentous fungi (Juge et al. 1998; Punt et al. 1987).
The gpd genes have also been cloned from basidiomycetous fungi, including Schizophyllum commune, Phanerochaete chrysosporium, Agaricus bisporus (Harmsen et al. 1992), and Lentinula edodes (Hirano et al. 1999). Among these mushrooms, genetic transformation using homologous gpd promoters was reported successful only in A. bisporus and L. edodes (Hirano et al. 2000; Van de Rhee et al. 1996). Although heterologous promoters have previously been used for the expression of drug-resistant marker genes, the genetic transformation was not sufficient to express heterologous genes (Ruiz-Diez 2002). To sufficiently and effectively express a heterologous gene, it is important for a host cell to recognize the promoter sequence by its transcriptional machinery.
In order to construct efficient transformation vectors allowing high-level expression of various heterologous genes in mushrooms, it is necessary to isolate a highly active promoter and use this promoter to develop a genetic transformation system. Most protocols used in fungal transformation involved electroporation of protoplasts (Chakraborty et al. 1991; Robinson and Sharon 1999; Van de Rhee et al. 1996; Ward et al. 1989), treatment with CaCl2 and polyethylene glycol (Ogawa et al. 1998; Sato et al. 1998), or restriction enzyme-mediated integration (Hirano et al. 2000; Irie et al. 2003; Sato et al. 1998). Since these transformation systems mainly relied on troublesome protoplast preparation, they were not easy to apply to all edible mushrooms, yielding insufficient regenerable protoplasts. Agrobacterium tumefaciens-mediated transformation is routinely used for the genetic modification of a wide range of plant species and demonstrates the ability to transfer DNA from a prokaryote to filamentous fungi (Chen et al. 2000; Combier et al. 2003; De Groot et al. 1998; Dunn-Coleman and Wang 1998; Mikosch et al. 2001). However, this method is not necessarily appropriate for all mushroom species.
In this study, we cloned the gpd gene from Flammulina velutipes, one of the most important edible mushrooms in Asia, and analyzed its complete nucleotide sequence, including the 5′ flanking region. We also developed a simple and reliable electroporation protocol for basidiospores. Using this protocol and the homologous gpd promoter, we demonstrated that a heterologous gene, the hygromycin B phosphotransferase gene (hph), can be successfully expressed in F. velutipes and this protocol can be applied to most mushroom species without special equipment or techniques.
Materials and methods
Strains and media
F. velutipes BCRC 37086 was purchased from the Bioresources Collection and Research Center (Hsinchu, Taiwan) and grown in either potato dextrose agar (PDA; Difco, Detroit, Mich.) or potato dextrose broth (PDB; Difco) at 25°C. Transformants were selected on PDA with 30 μg/ml of hygromycin (A.G. Scientific, San Diego, Calif.). Escherichia coli strain DH5α (GIBCO–BRL Life Technologies, Grand Island, N.Y.) was used for DNA manipulations and grown in LB medium (Sigma Chemical Co., St Louis, Mo.) at 37°C. PCR-amplified DNA fragments were purified, cloned into a pGEM-T Easy vector (Promega, Madison, Wis.), and sequenced.
Cloning of the gpd gene
Two degenerate primers, 5′-GKATCGGMCGYMYYGTMYYCMGHAATGC-3′ (corresponding to RIGRIVLRNA) and 5′-RTANCCCCAYTCRTTRTCRTACCA-3′ (corresponding to WYDNEWGY), were designed according to the conserved amino acid sequences of published basidiomycetous gpd genes (GenBank accession nos. BAA83550, AAA33926, AAA33732, AAA32634, AAA32633; Fig. 1). Using these degenerate primers and F. velutipes genomic DNA as the template, partial gpd of F. velutipes was cloned by PCR amplification.

Alignment of the deduced amino acid sequences of the GPD proteins of F. velutipes, L. edodes, Agaricus bisporus, P. chrysosporium, and S. commune. Identical residues are highlighted. Two conserved regions for degenerate oligonucleotide primers design are underlined. The main catalytic amino acid residue cysteine at position 151 is indicated by an asterisk
The 5′ flanking region and cDNA of F. velutipes gpd was cloned by means of the Clontech Universal GenomeWalker kit and SMART RACE cDNA amplification kit, according to the manufacturer’s instructions, respectively (BD Bioscience, Paolo Alto, Calif.). A gene-specific primer (GSP) was designed from the partial F. velutipes gpd sequence.
Plasmid construction
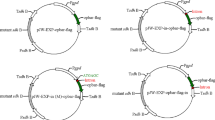
The hygromycin B phosphotransferase (hph) gene with cauliflower mosaic virus (CaMV) 35S terminator was amplified from pCAMBIA 1300 (CAMBIA, Canberra, Australia) using primers Hyg-f (5′-ACTAGTATGAAAAAGCCTGAACTCACC-3′) and Hyg-r (5′-CTGCAGACAACTTAATAACACATTGCG-3′). Then, the amplified fragment was cloned into a pGEM-T Easy vector and a 1.4-kb promoter region of the F. velutipes gpd gene was introduced to drive hph gene expression. The resulting plasmid, pFGH, was used for the transformation experiments.
Transformation procedure
Exponential-decay high-voltage electric pluses were delivered by BTX ECM 630 and 0.2-cm cuvettes (BTX, San Diego, Calif.). The electric pulse delivery test used several settings: capacitor 25 μF, resistance from 100 Ω to 800 Ω, and field strength from 6.25 kV/cm to 12.5 kV/cm.
Basidiospores were collected from F. velutipes fruit bodies, suspended in PDB, and then incubated overnight with gentle shaking at 25°C. These germinated basidiospores were harvested by centrifugation at 2,000 g for 5 min and resuspended in P buffer (0.02 M phosphate buffer, pH 5.8, 0.6 M mannitol) containing 2 mg/ml lysing enzymes (Sigma). After incubation for 2 h, these basidiospores were washed free of enzyme and transferred to a small volume of electroporation buffer (1 mM Hepes, pH 7.5, 0.6 M mannitol). Basidiospores (107–108) were mixed with 10 μg plasmid DNA, chilled on ice for 10 min, and subjected to electroporation. After pulse delivery, basidiospores were kept for 10 min on ice and mixed with PDB containing 0.6 M mannitol. Transformants were selected on PDA plates containing 30 μg/ml hygromycin.
Detection of introduced sequence in transformants and stability test
Genomic DNA isolated from putative hygromycin-resistant transformants was analyzed by PCR. Amplification of hph gene was carried out using primers Hyg-f and Hyg-r, previously used in plasmid construction. The amplified fragments were further identified by restriction enzyme Xho I digestion. Randomly selected transformants were transferred to medium without antibiotic selection for weeks to months, followed by a hygromycin resistance test.
Southern hybridization
Approximately 5 μg of genomic DNA digested with restriction enzymes were size-fractionated by electrophoresis on a 1% agarose gel; and the DNA fragments in the agarose gel were transferred to a Hybond N+ Nylon membrane (Amersham, Hong Kong) using 10× SSC. A genomic DNA fragment amplified by PCR with two gpd gene-specific primers (5′-GATCGGCCGTCTTGTCCTCC-3′, 5′-GCAATTGGTAGTGCAAGAAGCG-3′) was used as a subsequent probe for gpd. The DNA fragment amplified by PCR with primers Hyg-f and Hyg-r from pCAMBIA 1300 was used as a subsequent probe for transformants. Labeling of the DNA probe, hybridization, and signal detection were conducted by means of the digoxigenin (DIG)-probe synthesis and detection kit (Roche, Mannheim, Germany), according to the manufacturer’s instructions.
Results
Isolation and structure of F. velutipes gpd
Using degenerate primers designed according to conserved amino acid sequences of published basidiomycetous gpd genes, a fragment approximately 1.2 kb in size was amplified by PCR from the F. velutipes genomic DNA template. The 5′ flanking region of the F. velutipes gpd gene was amplified by genome walking and the full-length gpd cDNA of F. velutipes was obtained by aligning sequences obtained by 5′RACE and 3′RACE.
Based upon the alignment of cDNA sequences and a comparison with the genomic DNA sequence, the complete gpd sequence (from ATG to TAA) of F. velutipes is 1,489 bp in length, with the presence of nine introns (DDBJ/EMBL/GenBank nucleotide sequence database, accession number AF515622). The positions of introns were highly conserved in the gpd genes of the investigated basidiomycetes (Harmsen et al. 1992), especially A. bisporus (data not shown), which might reflect the evolutionary divergence of these mushrooms.
The GPD protein of F. velutipes is encoded by 339-amino acids, and the amino acid sequence is similar to that of other species (Fig. 1). The main catalytic amino acid residue responsible for the binding of glyceraldehyde-3-phosphate within the F. velutipes GPD protein is found at the 151st residue, cysteine. This observation was also found in other basidiomycetes including L. edodes, A. bisporus, P. chrysosporium, and S. commune (Hirano et al. 1999).
The sequence of the 5′ flanking region
There was one typical TATA box (TATAAAA) and two CAAT boxes (CCAAT) located in the 5′ flanking region. The CAAT boxes were located further upstream from the initiating ATG codon than in other published basidiomycetous gpd genes (Harmsen et al. 1992; Hirano et al. 1999). A pyrimidine region composed of stretches of thymine nucleotides interrupted by cytosine residues was observed immediately upstream of the transcription initiation site. Similar to other basidiomycetes (Harmsen et al. 1992), those consensus-promoter elements labeled as the gpd box, pgk box, qut box, and qa box in the A. nidulans and A. niger gpd promoter regions (Punt et al. 1990) were not found in the promoter region of the F. velutipes gpd gene.
Copy number of the gpd gene
Southern hybridization was conducted in order to detect the copy number of the gpd gene in the F. velutipes genome. Digested F. velutipes genomic DNA fragments were hybridized with a DIG-labeled genomic DNA fragment amplified by PCR using two gpd gene-specific primers. The expected specific gpd signals were detected following Southern hybridization (Fig. 2), suggesting that there is only one copy of gpd gene in the genome of F. velutipes. A similar result was also found in L. edodes (Hirano et al. 1999).

Southern blot analysis of genomic DNA and an illustration of the gpd gene. Numbers indicate the positions of restriction enzyme cutting sites for the gpd gene. A 600-bp genomic DNA fragment amplified by PCR using two gpd gene-specific primers was used as a probe. Xh Xho I, St Stu I, Nd Nde I, Bg Bgl II, Av Ava I, Ac Acc I
Transformation of F. velutipes basidiospores
Basidiospores after transformation were colonized on PDA with and without 30 μg/ml of hygromycin, respectively. The transformation efficiencies ranged over 5–60 transformants/μg DNA at all electric pulse delivery settings. This was in the same rank as results obtained by the REMI method (Hirano et al. 2000), but avoided protoplast preparation.
Subculturing transformants on media without selection pressure and followed by a hygromycin resistance test demonstrated that the hygromycin resistance trait was maintained stably during mitotic cell division for 3 months (Fig. 3). The gpd promoter of F. velutipes constitutively drove the hygromycin resistance trait, indicating the promise of downstream gene expression. The presence of hph DNA introduced via transformation was checked by PCR amplification and identified by Xho I digestion of the PCR-amplified fragments (Fig. 4). No false positives were detected by PCR amplification among 30 antibiotic-resistant cultures. In order to investigate the fate of transforming DNA, Southern blot analysis was performed for transformants (Fig. 5). Genomic DNA from transformants was digested with EcoR I and hybridized with a DIG-labeled hph gene probe. There is only one EcoR I cutting site within the plasmid pFGH and therefore hybridization bands could be used to determine the copy number of hph per genome. Figure 5 shows that the hph gene integrated one or three copies in tested transformants; and bands of various sizes were visualized. This result suggested that the introduced fragment was integrated randomly into the F. velutipes genome.

Transformants expressed the hygromycin resistance trait stably under a non-selective environment. Randomly selected transformants were subcultured on medium without hygromycin for 3 months and then transferred to PDA plates containing 30 μg/ml of hygromycin. WT Wild-type non-transformed parental strain

a Organization of pFGH. pFGH is 5.7 kb in size and consists of a pGEM-T Easy vector backbone containing the ampicillin resistance gene [Amp (R)]. The hygromycin resistance gene [Hygromycin (R)], with the CaMV35S terminator (CaMV 35S-3′), was joined to the F. velutipes gpd promoter (pGPD). Shown are restriction enzyme sites with map distance in kilobases. b PCR analysis of DNA isolated from putative hygromycin-resistant transformants. PCR amplification was carried out on genomic DNA using primers Hyg-f and Hyg-r defining a ca. 1.3-kb fragment containing the hph gene and CaMV 35S terminator. Lanes 1–8 DNA from randomly selected putative transformants, lane 9 negative control with water, lane 10 DNA isolated from non-transformed F. velutipes, lane 11 positive control with plasmid pFGH. c Identification of PCR-amplified fragments from b. Lanes 1–4 amplified fragments from lanes 1–4 of b, Lane 11, positive control with plasmid pFGH from lane 11 from b. a, b Respectively, treated without or with restriction enzyme XhoI, M DNA molecular size markers (kilobases)

Southern blot analysis of transformants. Lanes 1–4 EcoRI-digested genomic DNA of transformants probed with the DIG-labeled hph sequence, lane 5 positive control of pFGH
Discussion
F. velutipes gpd
By comparison of the DNA and cDNA sequences obtained above, the coding region of genomic gpd DNA was determined. It agreed perfectly with the cDNA clone, suggesting this gpd gene encodes a functional protein. Some fungi, such as Mucor circinelloides and A. bisporus, which harbor more than one gpd gene, contained only one gpd mRNA, indicating that there is only one functional gpd (Harmsen et al. 1992; Wolff and Arnau 2002). It is thus plausible to assume that the gpd isolated from F. velutipes should be a functional gene. The result of Southern analysis suggested that there is only one copy of the gpd gene in the genome of F. velutipes. In the case of Nde I and Acc I, there should be two positive bands rather than one. This might be due to incomplete digestion and non-optimal hybridization stringency. The ability of the promoter obtained in this study to drive heterologous gene expression was proved by the hygromycin resistance trait of transformants; and this indicated the promise of other downstream gene expression. To determine which part of the gpd promoter region is essential for its function, deletion analysis of shortened gpd promoter will start in the future.
Transformation of F. velutipes basidiospores
The results indicated that electroporation of basidiospores could be a useful method for basidiomycete transformation and that the gpd promoter of F. velutipes was useful in the expression of foreign genes. Our results also showed that the cell wall acts as a powerful barrier to the uptake of DNA during electroporation; and this is consistent with observations in other filamentous fungi (Chakraborty et al. 1991). The mild pretreatment of germinated basidiospores with lysing enzymes proved useful, since it did not seriously compromise the integrity of the cell wall or the viability of basidiospores, while it elicited a marked enhancement in the yield of transformants.
The use of basidiospores instead of vegetative mycelium as recipient provided an easy way to determine transformants without the false positives caused by tissue tolerance toward antibiotics and made the electroporation procedure more applicable. The variation in the copy number and position of hph between transformants indicated the integration of gene was a random event and might occur by non-homologous recombination (Fig. 5). No clear relationship between the diversity of integrated DNA and morphological differences nor the hph copy number and growth rate was found in this study. This came as no surprise, since each monokaryon germinated from a basidiospore has a different genomic background, consequently leading to a different morphology (Brown and Casselton 2001; Larraya et al. 2001). One additional advantage of using basidiospores in transformation is the ease of tracing heterologous genes, since the transformants are monokaryons instead of heterokaryons.
Although Agrobacterium tumefaciens-mediated transformation is useful in plants and some filamentous fungi, it does not work with F. velutipes, suggesting the species hedge might limit its extensive application. In addition, mushrooms are heterokaryons and it is easier to trace heterologous genes in transformants produced by basidiospore electroporation. The transformation procedure used in this study could be used in F. velutipes and other edible mushrooms, such as Agaricus bisporus and L. edodes without troublesome protoplast preparation, cocultivation, or expensive equipment. This procedure provides a tool for mushroom genetic research and makes molecular breeding a reality. This is the first report about transformation and expression of heterologous genes in the important edible mushroom F. velutipes.
References
Bitter GA, Egan KM (1988) Expression of interferon-gamma from hybrid yeast GPD promoters containing upstream regulatory sequences from the GAL1–GAL10 intergenic region. Gene 69:193–207
Brown AJ, Casselton LA (2001) Mating in mushrooms: increasing the chances but prolonging the affair. Trends Genet 17:393–400
Chakraborty BN, Patterson NA, Kapoor M (1991) An electroporation-based system for high-efficiency transformation of germinated conidia of filamentous fungi. Can J Microbiol 37:858–863
Chen X, Stone M, Schlagnhaufer C, Romaine CP (2000) A fruiting body tissue method for efficient Agrobacterium-mediated transformation of Agaricus bisporus. Appl Environ Microbiol 66:4510–4513
Combier JP, Melayah D, Raffier C, Gay G, Marmeisse R (2003) Agrobacterium tumefaciens-mediated transformation as a tool for insertional mutagenesis in the symbiotic ectomycorrhizal fungus Hebeloma cylindrosporum. FEMS Microbiol Lett 220:141–148
De Groot MJ, Bundock P, Hooykaas PJ, Beijersbergen AG (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat Biotechnol 16:839–842
Doring F, Klapper M, Theis S, Daniel H (1998) Use of the glyceraldehyde-3-phosphate dehydrogenase promoter for production of functional mammalian membrane transport proteins in the yeast Pichia pastoris. Biochem Biophys Res Commun 250:531–535
Dunn-Coleman N, Wang H (1998) Agrobacterium T-DNA: a silver bullet for filamentous fungi? Nat Biotechnol 16:817–818
Eriksson P, Andre L, Ansell R, Blomberg A, Adler L (1995) Cloning and characterization of GPD2, a second gene encoding sn-glycerol 3-phosphate dehydrogenase (NAD+) in Saccharomyces cerevisiae, and its comparison with GPD1. Mol Microbiol 17:95–107
Harmsen MC, Schuren FH, Moukha SM, Zuilen CM van, Punt PJ, Wessels JG (1992) Sequence analysis of the glyceraldehyde-3-phosphate dehydrogenase genes from the basidiomycetes Schizophyllum commune, Phanerochaete chrysosporium and Agaricus bisporus. Curr Genet 22:447–454
Hirano T, Sato T, Okawa K, Kanda K, Yaegashi K, Enei H (1999) Isolation and characterization of the glyceraldehyde-3-phosphate dehydrogenase gene of Lentinus edodes. Biosci Biotechnol Biochem 63:1223–1227
Hirano T, Sato T, Yaegashi K, Enei H (2000) Efficient transformation of the edible basidiomycete Lentinus edodes with a vector using a glyceraldehyde-3-phosphate dehydrogenase promoter to hygromycin B resistance. Mol Gen Genet 263:1047–1052
Holland MJ, Holland JP (1978) Isolation and identification of yeast messenger ribonucleic acids coding for enolase, glyceraldehyde-3-phosphate dehydrogenase, and phosphoglycerate kinase. Biochemistry 17:4900–4907
Irie T, Sato T, Saito K, Honda Y, Watanabe T, Kuwahara M, Enei H (2003) Construction of a homologous selectable marker gene for Lentinula edodes transformation. Biosci Biotechnol Biochem 67:2006–2009
Juge N, Svensson B, Williamson G (1998) Secretion, purification, and characterisation of barley alpha-amylase produced by heterologous gene expression in Aspergillus niger. Appl Microbiol Biotechnol 49:385–392
Larraya LM, Perez G, Iribarren I, Blanco JA, Alfonso M, Pisabarro AG, Ramirez L (2001) Relationship between monokaryotic growth rate and mating type in the edible basidiomycete Pleurotus ostreatus. Appl Environ Microbiol 67:3385–3390
Mikosch TS, Lavrijssen B, Sonnenberg AS, Griensven LJ van (2001) Transformation of the cultivated mushroom Agaricus bisporus (Lange) using T-DNA from Agrobacterium tumefaciens. Curr Genet 39:35–39
Ogawa K, Yamazaki T, Hasebe T, Kajiwara S, Watanabe A, Asada Y, Shishido K (1998) Molecular breeding of the basidiomycete Coprinus cinereus strains with high lignin-decolorization and -degradation activities using novel heterologous protein expression vectors. Appl Microbiol Biotechnol 49:285–289
Piechaczyk M, Blanchard JM, Marty L, Dani C, Panabieres F, El Sabouty S, Fort P, Jeanteur P (1984) Post-transcriptional regulation of glyceraldehyde-3-phosphate-dehydrogenase gene expression in rat tissues. Nucleic Acids Res 12:6951–6963
Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, Hondel CA van den (1987) Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124
Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, Hondel CA van den (1990) Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93:101–109
Robinson M, Sharon A (1999) Transformation of the bioherbicide Colletotrichum gloeosporioides f.sp. Aeschynomene By electroporation of germinated conidia. Curr Genet 36:98–104
Ruiz-Diez B (2002) Strategies for the transformation of filamentous fungi. J Appl Microbiol 92:189–195
Sato T, Yaegashi K, Ishii S, Hirano T, Kajiwara S, Shishido K, Enei H (1998) Transformation of the edible basidiomycete Lentinus edodes by restriction enzyme-mediated integration of plasmid DNA. Biosci Biotechnol Biochem 62:2346–2350
Van de Rhee MD, Graca PM, Huizing HJ, Mooibroek H (1996) Transformation of the cultivated mushroom, Agaricus bisporus, to hygromycin B resistance. Mol Gen Genet 250:252–258
Vassileva A, Chugh DA, Swaminathan S, Khanna N (2001) Expression of hepatitis B surface antigen in the methylotrophic yeast Pichia pastoris using the GAP promoter. J Biotechnol 88:21–35
Ward M, Kodama KH, Wilson LJ (1989) Transformation of Aspergillus awamori and A. niger by electroporation. Exp Mycol 13:289–293
Wolff AM, Arnau J (2002) Cloning of glyceraldehyde-3-phosphate dehydrogenase-encoding genes in Mucor circinelloides (syn. racemosus) and use of the gpd1 promoter for recombinant protein production. Fungal Genet Biol 35:21–29
Acknowledgements
The financial support of the National Science Council of the ROC (Grant No. NSC91-2317-B-002-040) is greatly appreciated. We thank Dr. Ruey-Shyang Hseu (Institute of Microbiology and Biochemistry, National Taiwan University) for his valuable scientific advice and encouragement associated with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kuo, CY., Chou, SY. & Huang, CT. Cloning of glyceraldehyde-3-phosphate dehydrogenase gene and use of the gpd promoter for transformation in Flammulina velutipes . Appl Microbiol Biotechnol 65, 593–599 (2004). https://doi.org/10.1007/s00253-004-1635-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1635-1