
Abstract
The gene encoding l-lactate dehydrogenase from Thermoanaerobacterium saccharolyticum JW/SL-YS485 was cloned, sequenced, and used to obtain an l-ldh deletion mutant strain (TD1) following a site-specific double-crossover event as confirmed by PCR and Southern blot. Growth rates and final cell densities were similar for strain TD1 and the wild-type grown on glucose and xylose. Lactic acid was below the limit of detection (0.3 mM) for strain TD1 on both glucose and xylose at all times tested, but was readily detected for the wild-type strain, with average final concentrations of 8.1and 1.8 mM on glucose and xylose, respectively. Elimination of lactic acid as a fermentation product was accompanied by a proportional increase in the yields of acetic acid and ethanol. The results reported here represent a step toward using metabolic engineering to develop strains of thermophilic anaerobic bacteria that do not produce organic acids, and support the methodological feasibility of this goal.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thermoanaerobacterium sp. strain JW/SL-YS485, a thermophilic, anaerobic, Gram-positive bacterium, was originally isolated from Yellowstone National Park, Wyoming (Liu et al. 1996), and subsequently designated Thermoanaerobacterium saccharolyticum JW/SL-YS485 (Mai and Wiegel 2000). This organism ferments xylan, but not cellulose, and produces ethanol, acetate, lactic acid, CO2, and H2 as fermentation products. T. saccharolyticum is of interest because of its role in carbohydrate utilization in high-temperature environments and its potential utility for production of ethanol from biomass. In particular, the pentose-fermenting capability of this organism might be used to complement a cellulose-fermenting thermophile such as Clostridium thermocellum, in a consolidated bioprocessing configuration featuring fermentation of lignocellulosic biomass in the absence of added saccharolytic enzymes (Lynd et al. 2002). Mai et al. (1997) have previously reported development of an electrotransformation protocol for T. saccharolyticum based on the shuttle vector pIKM1 with selection based on kanamycin resistance, and have used this protocol in conjunction with homologous recombination to express a cellobiohydrolase encoded by cbhA of C. thermocellum (Mai and Wiegel 2000).
Metabolic engineering of end-product metabolism has been pursued extensively in Escherichia coli, resulting in strains of industrial interest that produce high yields of ethanol (Ingram et al. 1998, 1999; Tao et al. 2001) as well as other products (Altaras and Cameron 1999; Causey et al. 2003; Nakamura et al. 2000; Nie et al. 2002; Vemuri et al. 2002; Zhou et al. 2003). However, such metabolic engineering has been pursued only to a limited extent in gram-positive, mesophilic, obligate anaerobes, and has not been reported for Gram-positive obligate anaerobic thermophiles. Expression of pyruvate decarboxylase and alcohol dehydrogenase in Clostridium cellulolyticum resulted in shifts in the distribution of end-products, although the yield of ethanol did not increase (Guedon et al. 2002). Butyrate kinase and phosphotransacetylase knockouts in Clostridium acetobutylicum were confirmed genetically but did not eliminate production of butyrate and acetate (Green et al. 1996), perhaps due to the presence of alloenzymes. Antisense RNA complementary to phosphotransbutyrylase and butyrate kinase genes has also been used in C. acetobutylicum to alter the yields of catabolic end-products and, although it increased solvent production, no decrease in acid formation was observed (Desai and Papoutsakis 1999).
In the work reported here, we sought to initiate investigation of metabolic engineering of end-product metabolism in T. saccharolyticum by knocking out the lactate dehydrogenase gene and thereby eliminating lactate production.
Materials and methods
Culture origin, maintenance, and cultivation
Thermoanaerobacterium saccharolyticum JW/SL-YS485 (DSM 8691) was kindly provided by Dr. J. Wiegel (University of Georgia, Athens, GA). Single colonies were picked and stored in 25% glycerol and 75% growth medium at −80 °C in 5-ml sealed serum vials under a N2 atmosphere. Cultures recovered from glycerol stocks were grown in liquid medium prior to use in experiments. Cultures were grown at 55 °C in DSM 61 broth (http://www.dsmz.de/media/med061.htm) supplemented with 0.5 g l-cysteine/l as reducing agent or in MYE medium containing mineral medium as described by Wiegel et al. (1979) supplemented with 2 g yeast extract/l. Xylose or glucose (5 g/l) was added as the growth substrate. DSM medium 61 was used for electrotransformation experiments, and MYE medium for strain characterization experiments. Growth on plates was performed at 55 °C in an anoxic chamber (Coy, Ann Arbor, Mich., USA) using DSM medium 61 or MYE medium with 2% agar. For selection of antibiotic-resistant strains, the medium was supplemented with 50–400 μg kanamycin/ml as noted in the text, and the culture temperature was maintained at 50 °C.
Plasmids and PCR primers
Plasmid pIKM1 (Mai et al. 1997), kindly provided by Dr. J. Wiegel, was used as the source of aph conferring thermostable kanamycin resistance (Trieu-Cout and Courvalin 1983) and as a positive control for gene transfer because it contains a thermostable Gram-positive origin of replication. Dr. J. Wiegel also provided a λ-ZAP (Stratagene, La Jolla, Calif., USA) genomic DNA library of T. saccharolyticum. Cloning vectors were obtained from commercial suppliers: TOPO pCR2.1 (Invitrogen, Carlsbad, Calif., USA); pUC18 (Invitrogen); pBLUESCRIPT II SK(+) (Stratagene); and pGEM-T (Promega, Madison, Wis., USA). PCR primers were synthesized by IDT (Coralville, Iowa, USA) and were designed based on sequences internal to either l-ldh of T. saccharolyticum (initialed L) or the aph kanamycin marker (initialed K). Primers used in this work were:
L-531–5′ACATGCATGCCTAACTCCAAGGAAACGAATAG3′
L′475–5′AACCTGCAGCAGTCCTTGCTTAAAAGGTATC3′
L527–5′GCTCTAGACAGAGTTTGCAGCATGGAGC3′
L′959–5′CGGGATCCGAAGCCTCCCAACATAATTTG3′
L-560–5′CGATAAAGAACATGGCTGG3′
L′2425–5′AAGAGGCCTCCACCACACATATG3′
K672–5′TATATAGAAGATATCGCCGTATGTAAGGAT3′
K′1824–5′CTAAATCTAGGTACTAAAATTCATCCA3′
K′805–5′GGTTATTGTCCTGGGTTTCAAGC3′
K1742–5′GACTTACTGGGGATCAAGCCTG3′
DNA preparation, manipulation, and Southern blot hybridization
DNA fragments were subcloned using standard protocols (Sambrook and Russell 2001). E. coli TOP10 (Invitrogen) was grown in Luria-Bertani medium supplemented with ampicillin (100 μg/ml) or kanamycin (50 μg/ml). Plasmid preparation and manipulation, genomic DNA preparation (Qiagen, Valencia, Calif., USA), transformation and hybridization (North2South, Pierce Biochemicals, Rockford, Ill., USA) were carried out using standard procedures or the suppliers’ instructions. Restriction enzymes were obtained from New England Biolabs (Beverly, Mass., USA), pfu from Stratagene and Taq polymerase from Promega. DNA sequencing reactions were conducted using the ABI BigDye terminator kit v3.0 (ABI, Foster City, Calif., USA) at the Dartmouth College Molecular Biology Core Facility (Hanover, N.H., USA).
Cloning of l-lactate dehydrogenase
Conserved regions of l-ldh genes from C. acetobutylicum ATCC824, Bacillus megaterium and Bacillus stearothermophilus ATCC12016 were identified as KPGETR, NPVDIL, GEHGD, and KGATYY; degenerate primers were designed with the CODEHOP program (Rose et al. 1998). l-ldh fragments within the portion of the gene bounded by conserved KPGETR and KGATYY regions were amplified using degenerate primers with T. saccharolyticum genomic DNA as the template and pfu as the DNA polymerase. A small fragment of ldh between the Sau3AI restriction sites was cloned from the genomic DNA library; the sequence matched the sequence of the corresponding fragment amplified with degenerate primers. The DNA fragments flanking the above-described portion of l-ldh were sequenced directly from genomic DNA using ThermoFidelase (Fidelity Systems, Gaithersburg Md., USA) with the BigDye Terminator kit v3.0. The sequence of the entire gene was compiled and deposited with GenBank as follows: T. saccharolyticum, accession number JW/SL-YS485-AY278026.
Knockout vector construction
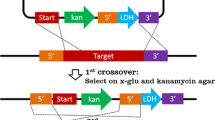
The 5.5-kb suicide vector pSGD8 (Fig. 1) was based on pUC18 using a design approach similar to that reported earlier (Mai and Wiegel 2000). l-ldh fragments ldh-up (1 kb) and ldh-down (0.43 kb) were amplified from genomic DNA using primer pairs l-531-L′475 and L527-L′959, respectively. PCR amplification was performed with pfu DNA polymerase. The resulting PCR fragments were A-tailed with Taq polymerase and cloned into TOPO pCR2.1 thereby facilitating the subcloning of the fragments. Following standard subcloning procedures, construct pSGD8 was obtained with the kanamycin marker aph flanked by ldh-up and ldh-down fragments. Restriction analysis and DNA sequencing confirmed the presence and orientation of the flanking regions and the aph marker.

Construction of pUC18 based Thermoanaerobacterium saccharolyticum l-ldh knockout vector pSGD8. Km, aph from shuttle vector pIKM1 conferring kanamycin resistance; Ap, bla from pUC18 conferring ampicillin resistance; ldh-up and ldh-down, partial ldh fragments. Nucleotides are numbered with respect to the ATG start site of ldh
Transformation of T. saccharolyticum
T. saccharolyticum was transformed using electroporation of autoplast-cell preparation, and kanamycin selection as described previously (Mai et al. 1997).
HPLC analysis
Glucose, xylose, acetate, lactate, and ethanol were analyzed by HPLC using an Aminex 87H column (Bio-Rad Laboratories, Hercules, Calif., USA) at 55 °C. The mobile phase consisted of 5 mM sulfuric acid at a flow rate of 0.7 ml/min. Detection was via refractive index using a Waters 410 refractometer (Milford, Mass., USA). The minimum detection level for lactate was 0.3 mM.
Calculation of carbon and electron recoveries
Carbon recovery was calculated exclusive of cell synthesis based on yields of organic fermentation products plus stoichiometrically associated CO2 production accompanying the production of ethanol and acetic acid (Lynd et al. 2002) using the formula:
where Y i/S is the yield of product i (mM product i/mM substrate; E for ethanol, A for acetic acid, and L for lactic acid) and N C/S is mM carbon/mM substrate (5 for xylose, 6 for glucose).
Recovery of available electrons was calculated exclusive of cell synthesis based on measured yields of organic fermentation products together with stoichiometrically associated production of H2 accompanying production of acetic acid (Lynd et al. 2002). Since there are 12 available electrons per mol ethanol, lactic acid, or acetic acid/H2 produced, and there are 20 available electrons per mol xylose and 24 available electrons per mol glucose (Papoutsakis 1984), available electron recovery is numerically equal to the carbon recovery as calculated using Eq. 1.
Results
Cloning of l-lactate dehydrogenase
The l-lactate dehydrogenase gene of T. saccharolyticum JW/SL-YS485 was cloned and sequenced using degenerate primers and direct sequencing of genomic DNA. Figure 2A shows the map of the sequenced l-ldh of T. saccharolyticum along with the location of various primers, ribosomal binding site (RBS), translation start and stop sites, and select DdeI restriction sites. The l-ldh ORF has similarity and identity relative to the l-ldh genes of other bacteria as follows: C. acetobutylicum ATCC824 (82%, 60%), B. megaterium (70%, 49%) and B. stearothermophilus 12016 (69%, 47%). A putative RBS rich in GC is located 7 bp upstream of the ATG start codon.

T. saccharolyticum l-ldh. RBS Putative ribosomal binding site. B Map of double crossover region with Δldh and inserted KmR gene. * Translational stop codon. Nucleotides are numbered with respect to the ATG start site of ldh
Chromosomal integration and isolation of strain TD1 with a deletion in l-ldh
T. saccharolyticum was transformed with the l-ldh knockout construct pSGD8 as well as replicative plasmid pIKM1. Electropulsed cell suspensions were recovered and cultured in selective liquid medium, with growth observed after 2 days in cultures transformed with pSGD8 and after 3 days in cultures transformed with pIKM1. Four presumptive chromosomal integrant isolates of T. saccharolyticum were picked from plates containing 50 µg kanamycin/ml streaked with cells transformed with pSGD8. All four isolates grew in liquid medium containing kanamycin at 75, 100, 200, and 400 μg/ml, and yielded identical results following PCR analysis and Southern blots (data not shown). In addition, none of the strains produced detectable lactic acid. One of the four isolates was chosen for further study and designated strain TD1.
Molecular characterization of strain TD1
PCR amplification using primers K672 and K′1824 and genomic DNA from T. saccharolyticum strain TD1 as template resulted in a fragment with the expected size of approximately 1.1 kb and confirmed the presence of the kanamycin gene (Fig. 3A, lane 1). PCR was also conducted using primer pairs consisting of one primer from the ldh gene that is not present in pSGD8 and one primer from the kanamycin gene. Primer pair l-560-K′805 yielded a 1.3-kb fragment (Fig. 3A, lane 2), which is the expected size for chromosomal integration of ldh-up (Fig. 2B). Primer pair K1742-L′2425 yielded a 0.7-kb fragment (Fig. 3A, lane 3), which was the expected size for a double-crossover event (Fig. 2B). All the above three PCR products were sequenced and confirmed. Negative controls with wild-type genomic DNA using the same PCR mix were conducted and none showed any PCR products (data not shown).

PCR analysis of l-ldh deletion mutant T. saccharolyticum strain TD1. Genomic DNA from strain TD1 was the template. Lane 1 Primers K672 and K′1824, lane 2 primers l-560 and K′805, lane 3 primers K1742 and L′2425. B Southern blot analysis of T. saccharolyticum wild-type and l-ldh deletion mutant TD1. Lane 1 Wild-type, lane 2 wild-type transformed with pIKM1, lane 3 deletion mutant TD1. DdeI genomic DNA digests were hybridized with the 0.43-kb ldh-down fragment as the probe
Further confirmation of the double-crossover event was obtained from Southern blot analysis. Genomic DNA from the wild-type, strain TD1, and from the wild-type strain transformed with plasmid pIKM1 was digested with DdeI, and the 0.43 kb ldh-down fragment was used as a probe for hybridization. Both wild-type strains hybridized to a 1.1-kb fragment (Fig. 3B, lanes 1 and 2), whereas the deletion mutant strain TD1 hybridized to a 1.4-kb fragment (Fig. 3B, lane 3). This confirmed that the chromosomal integration was in fact a double-crossover event because a single-crossover event would have resulted in a different fragment size.
Physiological characterization of strain TD1
Figure 4 presents representative batch growth curves for T. saccharolyticum wild-type and TD1 strains on glucose (Fig. 4A, B) and xylose (Fig. 4C, D). Two other replicate experiments showed similar trends (data not shown). The time required to achieve substrate exhaustion and the final optical densities achieved in stationary phase were very similar for the wild-type and deletion mutant strains growing on either glucose or xylose. Specific growth rates calculated during exponential phase were 0.4±0.03 h−1 for the wild-type on both glucose and xylose and for strain TD1 on glucose, while the growth rate of strain TD1 on xylose was 0.3±0.02 h−1. Lactic acid was below the limit of detection (0.3 mM) for growth of strain TD1 on both glucose and xylose at all times tested, but was readily detected for growth of the wild-type on both glucose and xylose, with average final concentrations of 8.1 and 1.8 mM, respectively. After three transfers in batch culture in the absence of selective pressure, strain TD1 still did not produce lactic acid and did not show any major changes in the yields of ethanol or acetate, suggesting stable gene deletion.

Growth curves for T. saccharolyticum wild-type and TD1 strains on glucose and xylose. A Wild-type, glucose; B TD1, glucose; C wild-type, xylose; D TD1, xylose. Substrate (◯), lactate (●), acetate (Δ), ethanol (□) and OD (x)
Glucose consumption, fermentation product formation, product and cell yields, and carbon recovery are tabulated in Table 1 for batch cultures of strains T. saccharolyticum TD1 and wild-type, in pre-reduced MYE medium. Carbon recoveries were rather similar for the two strains but somewhat in excess of 100%. This excess may be due to the assimilation of components in yeast extract, which is present in the growth medium at a concentration of 2 g/l. Cell yields, evaluated in terms of change in OD per mM substrate consumed, were higher for strain TD1 than for the wild-type for both glucose and xylose as substrates, although the difference was only statistically significant for glucose (p= 0.1).
Discussion
The deduced amino acid sequence encoded by l-ldh of T. saccharolyticum JW/SL-YS485 exhibited significant similarity relative to previously sequenced l-ldh genes from mesophilic and thermophilic bacteria. The observation that three different degenerate primer pairs yielded the same ORF and single bands were obtained upon Southern blotting of genomic DNA confirm that l-ldh is present in a single copy in the chromosome of T. saccharolyticum.
T. saccharolyticum strain TD1 featuring a homologous recombination-mediated knockout of l-ldh was obtained with little difficulty. Data from PCR and Southern blots indicate that deletion of the single-copy l-ldh occurred via a double-crossover event. The growth rates of the wild-type strain and the deletion mutant strain TD1 are in the range reported by other researchers for growth at 55 °C (Mai and Wiegel 2000). Lactic acid production was not detectable in stain TD1 during growth on both glucose and xylose. Elimination of lactic acid as a fermentation product resulted in proportionately increased yields of acetic acid and ethanol in replicate experiments. No additional end-products were detected by HPLC in strain TD1 as compared to the wild-type strain, and the magnitude of the increased ethanol and acetate yields were sufficient to account for the decrease in carbon flow to lactic acid. Mat-Jan et al. (1989) previously examined end-product redistribution in a fermentative ldh mutant of E. coli. After 4 h of growth, they found no detectable amounts of lactate and a slight decrease in ethanol and acetate formation compared to the parent strain; however, during subsequent incubation substantial amounts of pyruvate accumulated.
The work reported here represents a step toward developing strains of thermophilic anaerobic bacteria that do not produce organic acids. Such strains are doubly desirable in order to pursue advances in conversion of lignocellulosic biomass to ethanol. In addition to increasing ethanol yield, strains in which organic acid production has been eliminated also provide a means to test the hypothesis that higher concentrations of ethanol can be produced when cells are not subject to inhibition by organic acids and their salts (Lynd et al. 2001, 2002).
References
Altaras NE, Cameron DC (1999) Metabolic engineering of a 1,2-propanediol pathway in Escherichia coli. Appl Environ Microbiol 65:1180–1185
Causey TB, Zhou S, Shanmugam KT, Ingram LO (2003) Engineering the metabolism of Escherichia coli W3110 for the conversion of sugar to redox-neutral and oxidized products: Homoacetate production. Proc Natl Acad Sci USA 100:825–832
Desai RP, Papoutsakis ET (1999) Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl Environ Microbiol 65:936–945
Green EM, Boynton ZL, Harris LM, Rudolph FB, Papoutsakis ET, Bennett GN (1996) Genetic manipulation of acid formation pathways by gene inactivation in Clostridium acetobutylicum ATCC 824. Microbiology 142:2079–2086
Guedon E, Desvaux M, Petitdemange H (2002) Improvement of cellulolytic properties of Clostridium cellulolyticum by metabolic engineering. Appl Environ Microbiol 68:53–58
Ingram LO, Gomez PF, Lai X, Moniruzzaman M, Wood BE, Yomano LP, York SW (1998) Metabolic engineering of bacteria for ethanol production. Biotechnol Bioeng 58:204–214
Ingram LO, Aldrich HC, Borges ACC, Causey TB, Martinez A, Morales F, Saleh A, Underwood SA, Yomano LP, York SW, Zaldivar J, Zhou SD (1999) Enteric bacterial catalysts for fuel ethanol production. Biotechnol Prog 15:855–866
Liu SY, Gherardini FC, Matuschek M, Bahl H, Wiegel J (1996) Cloning, sequencing, and expression of the gene encoding a large S-layer-associated endoxylanase from Thermoanaerobacterium sp strain JW/SL-YS 485 in Escherichia coli. J Bacteriol 178:1539–1547
Lynd LR, Baskaran S, Casten S (2001) Salt accumulation resulting from base added for pH control, and not ethanol, limits growth of Thermoanaerobacterium thermosaccharolyticum HG-8 at elevated feed xylose concentrations in continuous culture. Biotechnol Prog 17:118–125
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial cellulose utilization: Fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577
Mai V, Wiegel J (2000) Advances in development of a genetic system for Thermoanaerobacterium spp: Expression of genes encoding hydrolytic enzymes, development of a second shuttle vector, and integration of genes into the chromosome. Appl Environ Microbiol 66:4817–4821
Mai V, Lorenz WW, Wiegel J (1997) Transformation of Thermoaerobacterium sp Strain JW/SL-YS485 with plasmid pIKM1 conferring kanamycin resistance. FEMS Microbiol Lett 148:163–167
Mat-Jan F, Alam KY, Clark DP (1989) Mutants of Escherichia coli deficient in the fermentative lactate dehydrogenase. J Bacteriol 171:342–348
Nakamura CE, Gatenby AA, Hsu AK-H, La Reau RD, Haynie SL, Diaz-Torres M, Trimbur DE, Whited GM, Nagarajan V, Payne MS, Picataggio SK, Nair RV (January 2000) Method for the production of 1,3-propanediol by recombinant microorganisms. US Patent 6,013,494
Nie W, Draths KM, Frost JW (2002) Benzene-free synthesis of adipic acid. Biotechnol Prog 18:201–211
Papoutsakis ET (1984) Equations and calculations for fermentations of butyric-acid bacteria. Biotechnol Bioeng 26:174–187
Rose TM, Schultz, ER Henikoff JG, Pietrokovski S, McCallum CM, Henikoff S (1998) Consensus-degenerate hybrid oligonucleotide primers for amplification of distantly-related sequences. Nucleic Acids Res 26(7):1628–1635
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Tao H, Gonzalez R, Martinez A, Rodriguez M, Ingram LO, Preston JF, Shanmugam KT (2001) Engineering a homo-ethanol pathway in Escherichia coli: Increased glycolytic flux and levels of expression of glycolytic genes during xylose fermentation. J Bacteriol 183:2979–2988
Trieu-Cuot P, P Courvalin (1983) Nucleotide sequence of the S faecalis plasmid gene encoding the 3′5′′-aminoglycoside phosphotransferase type III. Gene 23:331–341
Vemuri GN, Eiteman MA, Altman E (2002) Effects of growth mode and pyruvate decarboxylase on succinic acid production by metabolically engineered strains of Escherichia coli. Appl Environ Microbiol 68:1715–1727
Wiegel J, Ljundahl LG, Lawson JR (1979) Isolation from soil and properties of the extreme thermophile Clostridium thermohydrosulfuricum. J Bacteriol 139:800–810
Zhou SD, Causey TB, Hasona A, Shanmugam KT, Ingram LO (2003) Production of optically pure d-lactic acid in mineral salts medium by metabolically engineered Escherichia coli W3110. Appl Environ Microbiol 69:399–407
Acknowledgements
We thank Dr. J. Wiegel for providing us with the wild-type strain Thermoanaerobacterium saccharolyticum JW/SL-YS485 and its genomic DNA library. The support of the National Institute of Standards and Technology (grant no. 60NANB1D0064) and the Link Foundation is gratefully acknowledged. The experiments reported in this study comply with the current laws of the country in which they were performed.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Desai, S.G., Guerinot, M.L. & Lynd, L.R. Cloning of l-lactate dehydrogenase and elimination of lactic acid production via gene knockout in Thermoanaerobacterium saccharolyticum JW/SL-YS485. Appl Microbiol Biotechnol 65, 600–605 (2004). https://doi.org/10.1007/s00253-004-1575-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-004-1575-9