
Abstract
A whole-cell biotransformation system for the conversion of d-fructose to d-mannitol was developed in Escherichia coli by constructing a recombinant oxidation/reduction cycle. First, the mdh gene, encoding mannitol dehydrogenase of Leuconostoc pseudomesenteroides ATCC 12291 (MDH), was expressed, effecting strong catalytic activity of an NADH-dependent reduction of d-fructose to d-mannitol in cell extracts of the recombinant E. coli strain. By contrast whole cells of the strain were unable to produce d-mannitol from d-fructose. To provide a source of reduction equivalents needed for d-fructose reduction, the fdh gene from Mycobacterium vaccae N10 (FDH), encoding formate dehydrogenase, was functionally co-expressed. FDH generates the NADH used for d-fructose reduction by dehydrogenation of formate to carbon dioxide. These recombinant E. coli cells were able to form d-mannitol from d-fructose in a low but significant quantity (15 mM). The introduction of a further gene, encoding the glucose facilitator protein of Zymomonas mobilis (GLF), allowed the cells to efficiently take up d-fructose, without simultaneous phosphorylation. Resting cells of this E. coli strain (3 g cell dry weight/l) produced 216 mM d-mannitol in 17 h. Due to equimolar formation of sodium hydroxide during NAD+-dependent oxidation of sodium formate to carbon dioxide, the pH value of the buffered biotransformation system increased by one pH unit within 2 h. Biotransformations conducted under pH control by formic-acid addition yielded d-mannitol at a concentration of 362 mM within 8 h. The yield Y D-mannitol/D-fructosewas 84 mol%. These results show that the recombinant strain of E. coli can be utilized as an efficient biocatalyst for d-mannitol formation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
d-Mannitol is used commercially in pharmaceutical products and in the food industry as a sweetener, also for diabetic confectionery. Its clinical applications include as an osmotic diuretic and an inert excipient for medical agents, while its derivative d-mannitol hexanitrate is used as a vasodilator (Johnson 1976). The annual production of d-mannitol is in the range of 40,000 tons. In the industrial large-scale production of d-mannitol, mixtures of glucose and fructose are reduced by catalytic hydrogenation on activated nickel catalysts. Due to the poor selectivity of the reaction, besides d-mannitol d-sorbitol is formed as a by-product in three-fold excess, thus requiring expensive downstream processing of the product (Makkee et al.1985; Soetaert et al. 1999). Therefore, during the past few years a number of investigations on enzymatic d-mannitol production have been carried out. Enzyme-catalyzed processes employing purified mannitol dehydrogenases from different sources in combination with a cofactor-regenerating system were shown to be far more efficient with respect to reaction selectivity and yield (Haltrich et al. 1996; Nidetzky et al. 1996; Slatner et al. 1998a, b). Utilizing purified mannitol dehydrogenase from Pseudomonas fluorescens and formate dehydrogenase from Candida boidinii, the batchwise reduction of d-fructose resulted in a final concentration of d-mannitol of 72 g/l and a fructose conversion of 80% (Slatner et al. 1998b). Furthermore, efforts have been made to establish microbial production systems for d-mannitol with heterofermentative lactic acid bacteria, e.g. Leuconostoc mesenteroides ATCC 9135 and L. pseudomesenteroides ATCC 12291. In these processes, mixtures of d-glucose and d-fructose were used as substrates, either with growing cultures or with resting cells in cell-recycle cultures (Soetaert et al. 1995; von Weymarn et al. 2002). About 95% of the initial d-fructose was converted to d-mannitol whereas d-glucose served as the carbon and energy source for growth and provided the reducing equivalents for reduction of d-fructose. Glucose was metabolized to lactic acid, acetic acid, and ethanol as by-products.
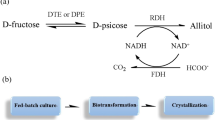
The present report describes a novel approach to setting up a recombinant oxidation/reduction cycle in Escherichia coli for d-mannitol formation. Formate dehydrogenase generates NADH from the intracellular NAD+-pool by oxidizing formate to volatile carbon dioxide, without any formation of further by-products. The NADH obtained is subsequently oxidized by an NAD-dependent mannitol dehydrogenase, thereby reducing d-fructose to d-mannitol. Introduction of a gene encoding a transporter for d-fructose enabled the cells to import d-fructose (Fig. 1). Resting batch and fed-batch cultures of this E. coli strain and strains expressing different combinations of recombinant genes were tested for their ability to form d-mannitol.

A recombinant oxidation/reduction cycle in Escherichia coli for d-mannitol formation
Material and methods
Bacterial strains, plasmids, media, and growth conditions
E. coli strains were transformed by the method described by Hanahan (1983) and cultivated in Luria-Bertani (LB) medium or on agar plates (Miller 1972). E. coli DH5α (Hanahan 1983) was used for cloning purposes and E. coli BL21 Star (DE3) (Invitrogen, Karlsruhe, Germany) for gene expression and whole-cell biotransformation. Plasmids were selected by adding antibiotics to the medium at a final concentration of 200 μg ampicillin/ml or 50 μg carbenicillin/ml (pLmdh, pMcFDH), 50 μg kanamycin/ml (pET24a(+), pET24Lmdj, pET24[fdhmdh]), and 25 μg chloramphenicol/ml (pZY507glf). For construction of recombinant E. coli harboring pET24[fdhmdh] and pZY507glf, pET24[fdhmdh] was first transformed. After selection of a positive transformant, competent cells were prepared and pZY507glf was co-transformed. The resulting transformants were selected for both plasmids. Basal expression from pET24a(+) vectors before IPTG induction was repressed by adding 0.4% (w/v) d-glucose. Expression from plasmid pZY507glf was regulated by a lacI q-tac promoter system and was also induced with IPTG (Weisser et al. 1995). Expression parameters were optimized for maximum formate and mannitol dehydrogenases activities with respect to inoculation, IPTG concentration, cell density at induction, and expression time. The different strains of recombinant E. coli BL21 (DE3) carrying any of the plasmids listed in Table 1, or combinations of them, were cultivated by inoculating a single colony of each strain into 20 ml LB medium with the appropriate additions as described above. These precultures were grown over-night at 30°C and 130 rpm and were used to inoculate the main cultures to an OD600 of 0.1. Main cultures were grown in LB medium in the presence of the appropriate additions (see above) at 30°C and 250 rpm to an OD600of 0.5, induced with 0.7 mM IPTG, and incubated at 27°C and 200 rpm for 4–5 h.
Plasmids pMcFDH and pZY507glf were the kind gifts of N. Esaki (Japan) and G.A. Sprenger (Germany), respectively.
Cloning and DNA techniques
For DNA manipulation, standard methods were used as described by Sambrook et al. (1989). Gene amplification and modification were done by standard PCR. mdh of Leuconostoc pseudomesenteroides was amplified from plasmid pLmdh with oligonucleotide primers forward 5′-CGAACATATGGAAGCACTTGTGTTAACTGGTAC –3′ and reverse 5′–ACAGCTCGAGTTATGCCTCTTCGCCACCAACC–3′. The resulting PCR product was cloned into pET24a(+) via a 5′ NdeI site and a 3′ XhoI site (italics) leading to plasmid pET24Lmdh (Hahn et al. 2003). Plasmid pET24[fdhmdh] was constructed by cloning mdh of L. pseudomesenteroides together with its wild-type ribosome binding site into pET24a(+) using a 5′ SacI and a 3′ XhoI site (italics) with oligonucleotide primers forward 5′-CGATGAGCTCAAAAGGAGAACAAACATGGAAGCACTTGTGTTAACT–3′ and reverse 5′-ACAGCTCGAGTTATGCCTCTTCGCCACCAACC-3′ in a PCR reaction with pLmdh vector DNA as template. After selection of a positive plasmid bearing mdh, fdh of Mycobacterium vaccae N10 was cloned upstream of mdh using a 5′ NdeI and a 3′ BamHI site (italics) with oligonucleotide primers forward 5′-CGATCATATGGCAAAGGTCCTGTGCGTTCTTTACGATGATCCG-3′ and reverse 5′-GCTAGGATCCTCAGACCGCCTTCTTGAACTTGGCGGCCTCTTC-3′ in a PCR reaction with pMcFDH vector DNA as template (Fig. 2). Oligonucleotides were obtained from MWG Biotech (Ebersberg, Germany). DNA was sequenced by SeqLab (Göttingen, Germany).

Genetic arrangement of the recombinant operon cloned into plasmid pET24a(+), containing the formate dehydrogenase gene from M. vaccae (fdh) and the mannitol dehydrogenase gene from L. pseudomesenteroides (mdh). lac O, lac operator; rbs, ribosome binding sites
Cell-free extract and enzyme assays
The cells were harvested by centrifugation (10,000 g and 4°C for 10 min), followed by a washing step with 100 mM potassium phosphate buffer, pH 6.5. Cells were disrupted by sonication (4 min, 4°C) and crude extracts were centrifuged at 20,000 g for 1 h at 4°C. The supernatants were used as cell-free extracts. The activity of formate dehydrogenase was determined photometrically in an assay mixture of 200 mM sodium formate, 2 mM NAD, and 100 mM potassium phosphate buffer, pH 7.0 (Schütte et al. 1976). The assay for mannitol dehydrogenase contained 200 μM NADH and 200 mM d-fructose in 100 mM potassium phosphate buffer, pH 6.0 (Hahn et al. 2003). One unit of enzyme activity (U) was defined as the amount of enzyme catalyzing the conversion of 1 μmol pyridine nucleotide per min at 30°C. Protein concentrations were determined by the method of Bradford (1976) using bovine serum albumin as the standard.
Whole-cell biotransformation
Cells from 400 ml culture medium (growth conditions, see above) were taken for each biotransformation. The cells were harvested by centrifugation (5,000 g for 5 min) and washed with 100 mM potassium phosphate buffer, pH 6.5. Twenty g cell wet weight/l (corresponding to 3 g cell dry weight/l) were resuspended in reaction solution containing 500 mM d-fructose and 500 mM sodium formate in 100 mM potassium phosphate buffer, pH 6.5. The reaction temperature was 30°C. For pH-static conditions, the suspension was incubated in a glass vessel with automatic pH control (titration with 3 M formic acid) at pH 6.5 with 120 rpm stirring. Incubation times were as indicated in the text. Samples of the biotransformation system were centrifuged, filtrated, and the resulting supernatants were analyzed by HPLC.
HPLC analysis
d-mannitol and d-fructose were quantified with a Biorad HPX-87C 300×7.8 mm column at 85°C and using H2O as the eluent. Substances were detected photometrically at 190 nm. Retention times for d-mannitol and d-fructose were 15.80 and 12.50 min, respectively. Formate was quantified on a Biorad HPX-87H 300 ×7.8 mm column at 65°C and using 6 mM H2SO4 as the eluent. Under these conditions, the retention time of formate was 12.80 min. Flow rates were 0.6 ml/min.
Nucleotide sequence accession numbers
The nucleotide sequences of the genes used in this work are available under GenBank accession numbers:mdh: AJ486977 (Hahn et al. 2003); fdh: AB072394 (Galkin et al. 1995); glf: M37982 (Barnell et al. 1990).
Results
E. coli pET24Lmdh, expressing mannitol dehydrogenase from Leuconostoc pseudomesenteroides
In order to engineer an E. coli strain containing mannitol dehydrogenase activity, strain E. coli pET24Lmdh was constructed carrying mdh, which encodes mannitol dehydrogenase from the heterofermentative lactic acid bacterium L. pseudomesenteroides (Hahn et al. 2003). Cell-free extracts of induced E. coli pET24Lmdh cultures exhibited a high specific mannitol dehydrogenase activity of 70 U/mg protein. However, whole cells of strain E. coli pET24Lmdh did not form d-mannitol when tested as catalysts in a biotransformation assay containing 500 mM each of formate and d-fructose in 100 mM potassium phosphate buffer, pH 6.5 (Table 2). This led to the conclusion that the strain was not able to provide a sufficiently high reduction state of the pyridine nucleotide pool to drive the MDH-catalyzed reduction of d-fructose to d-mannitol. Therefore, an enzyme-catalyzed, electron-donating, cofactor NADH regenerating reaction was implemented in strain E. coli pET24Lmdh.
E. coli pET24[fdhmdh], expressing mannitol dehydrogenase from L. pseudomesenteroides and formate dehydrogenase from Mycobacterium vaccae
For the construction of a recombinant oxidation/reduction cycle, the formate dehydrogenase-catalyzed, NAD+-dependent oxidation of formic acid to carbon dioxide was chosen as the cofactor regeneration system. fdh, encoding formate dehydrogenase from Mycobacterium vaccae N10 (Galkin et al. 1995), was first expressed with an activity of 1.0 U/mg cell-free protein in E. coli pMcFDH (Table 1). In order to construct E. coli pET24[fdhmdh] expressing formate dehydrogenase and mannitol dehydrogenase from a recombinant operon, mdh and fdh were amplified by PCR reactions with plasmid pLmdh and plasmid pMcFDH as templates and cloned successively into plasmid pET24a(+) (Fig. 2). In this recombinant operon the genes were transcribed via the T7 promoter and transcription was regulated by lac repressor binding to the lac operator. A T7 transcription terminator was located downstream of mdh. Translation of the transcribed genes was achieved using ribosome binding sites that originated from plasmid pET24a(+) for fdh, and from wild-type L. pseudomesenteroides for mdh. In expression studies with E. coli pET24[fdhmdh], cell-free extracts of induced cells had activities for FDH of 1.0 U/mg protein and for MDH of 14.0 U/mg protein. Biotransformation experiments with this strain yielded d-mannitol in concentrations of 15–20 mM from d-fructose and sodium formate, each at initial concentrations of 500 mM (Table 2). It was presumed that the low biotransformation efficiency of E. coli pET24[fdhmdh] was due to an insufficient import of free d-fructose into the cells. As in E. coli the uptake of d-fructose is catalyzed by phosphoenolpyruvate/ glucose phosphotransferase uptake systems (Kornberg 2001) a phosphoryl moiety from phosphoenolpyruvate is transferred to the sugar molecule, forming inside the cell either 1-phosphofructose (fructose II′BC-IIAMH) or 6-phosphofructose (mannose IIAB-IIC-IID) (Kornberg 2001).
E. coli pET24[fdhmdh] pZY507glf, expressing mannitol dehydrogenase from L. pseudomesenteroides, formate dehydrogenase from M. vaccae, and glucose facilitator protein from Zymomonas mobilis
To allow for efficient transport of free d-fructose into the E. coli cell, glf, encoding a glucose facilitator transport protein (GLF) of Zymomonas mobilis (Parker et al. 1995; Weisser et al. 1995) was additionally expressed in E. coli pET24[fdhmdh] by introducing plasmid pZY507glf. The resulting strain, E. coli pET24[fdhmdh] pZY507glf, showed MDH activities of 13.0 U/mg cell-free protein and FDH activities of 1.0 U/mg cell-free protein. In a biotransformation experiment this strain produced 216 mM d-mannitol, thus confirming the necessity of the presence of recombinant GLF (Table 2). Also essential was the presence of formate dehydrogenase catalyzing the electron-donating, cofactor NADH regenerating reaction, as could be seen from the comparison of biotransformation results of strains E. coli pET24[fdhmdh] pZY507glf (216 mM d-mannitol) and E. coli pET24Lmdh pZY507glf (20 mM d-mannitol). The inherent capacity of strain E. coli pET24Lmdh pZY507glf to reduce d-fructose to d-mannitol was only 10% of that of strain E. coli pET24[fdhmdh] pZY507glf (Table 2). In conclusion, the activities of formate and mannitol dehydrogenases and the d-glucose (d-fructose) transporter GLF are required for efficient operation of the recombinant oxidation/reduction cycle in E. coli cells.
Whole-cell biotransformations
Biotransformation without pH-control
E. coli pET24[fdhmdh] pZY507glf expressing all three recombinant genes, fdh, mdh and glf, appeared to be a promising biocatalyst for d-mannitol formation. Therefore, a more detailed study of the time course of d-mannitol formation by this E. coli strain was conducted (Fig. 3). Although the biotransformation was carried out in a 100 mM potassium phosphate buffer at pH 6.5, an increase of pH from 6.5 to 7.5 occurred during the first 2 h in this biotransformation system. The change of pH resulted from the formation of sodium hydroxide from sodium formate when formic acid was oxidized to carbon dioxide. After 8 h, about 210 mM of d-mannitol was formed from 230 mM d-fructose with a yield Y D-mannitol/D-fructose of 92 (mol%). Within the first 2 h, a high d-mannitol formation rate of 3.3 g·(g cell dry weight·h)–1 was observed, slowing down to 1.0 g·(g cell dry weight·h)–1 in the following time period (Fig. 3).

d-Mannitol formation (◆) by E. coli BL21 (DE3) pET24[fdhmdh] pZY507glf. □ d-Fructose consumption, ▲ pH. The time course of d-mannitol formation was determined by averaging the values of two independent biotransformations
Biotransformation with pH-control
To overcome the problem of increasing pH, the biotransformation was carried out under pH-static conditions. The pH was kept constant at 6.5, with formic acid serving at the same time as a substrate for FDH. Under this condition, efficient d-mannitol formation with high production rates of 4.1 g·(g cell dry weight·h)–1 was maintained during the first 3–4 h (Fig. 4). After that time the specific productivity decreased to 1.3 g·(g cell dry weight·h)–1. After 8 h, 362 mM d-mannitol had been formed from 433 mM d-fructose with a yield Y D-mannitol/D-fructose of 84 (mol%). Only 67 mM d-fructose of the initial 500 mM d-fructose was left in the reaction buffer. The activities of FDH and MDH remained constant in cell-free extracts of samples taken after 8 h and 17 h from the biotransformation.

d-Mannitol formation (◆) by E. coli BL21 (DE3) pET24[fdhmdh] pZY507glf. The biotransformation system contained 500 mM d-fructose and 250 mM sodium formate in 50 mM potassium phosphate buffer, pH 6.5. The pH value was kept constant by titration with 3 M formic acid. □ d-Fructose consumption, ▲ pH. The time course of d-mannitol formation was determined by averaging the values of four independent biotransformations
To achieve higher product concentrations a fed-batch of d-fructose was conducted in a further pH-static biotransformation. After 9 h, when the d-fructose concentration had decreased from 500 mM to 26 mM, 8 ml of a 3 M d-fructose solution were added to the biotransformation system (100 ml) to a final concentration of 260 mM. This resulted in a d-mannitol concentration of 500 mM and a residual d-fructose concentration of 100 mM after 23 h, thus pointing to the applicability of fed-batch techniques for this biotransformation process.
Discussion
A variety of whole-cell biotransformations with wild-type microorganisms have been developed, e.g. for the production of the chiral ephedrine-precursor (R)-phenylacetylcarbinol (Budesinsky and Protiva 1961; Bringer-Meyer and Sahm 1988; Rogers et al. 1997), steroid biotransformation (Murray and Petersen 1952; Mahato and Garei 1997) and regioselective oxidations of sugars and sugar alcohols with Gluconobacter oxydans (Reichstein and Grüssner 1934; Schedel 2000). With emerging recombinant DNA technology, biotransformation systems have been developed in which the genes encoding the desired enzymes are overexpressed in a host organism.
Industrially interesting products e.g. chiral alcohols and optically active amino acids synthesized from oxidized precursors require cofactors such as NADH and NADPH. Microbial conversions utilizing recombinant E. coli cells and different cofactor regeneration systems have been described (Endo and Koizumi 2001). The asymmetric reduction of ethyl 4-chloro-3-oxobutanoate to (R)-4-chloro-3-hydroxybutanoate was achieved by using resting cells of recombinant E. coli overexpressing an aldehyde reductase gene and a glucose dehydrogenase gene as catalyst (Kataoka et al.1997). l-Amino acids were produced from α-keto acids with E. coli overexpressing heterologous genes of l-amino acid dehydrogenase, alanine dehydrogenase, and formate dehydrogenase using the intracellular pool of NAD+ for the regeneration of NADH (Galkin et al. 1997). These examples demonstrate the potential of whole-cell biotransformations for manufacturing a great variety of compounds.
In the present work, d-mannitol production from d-fructose by means of reductive whole-cell biotransformation with resting cells of a recombinant strain of E. coli was shown to be an efficient process. Engineering of the production strain comprised the introduction of three plasmid-encoded genes, mdh, fdh and glf. Resting cells of strain E. coli pET24[fdhmdh], which lack the glucose facilitator gene, also carried out biotransformation of d-fructose and formate to d-mannitol and carbon dioxide, but produced only a low quantity of 15 mM d-mannitol. Since E. coli uses the phosphoenolpyruvate-dependent phosphotransferase system (PTS) for fructose uptake (Kornberg et al. 2000), the transport mechanism of d-fructose without phosphorylation into the cells was unclear. However, considering the high concentration of d-fructose (500 mM) in the biotransformation an unspecific transport of free fructose into the cell could be hypothesized for biotransformations with E. coli pET24[fdhmdh] as catalyst. The ethanologenic bacterium Zymomonas mobilis possesses a facilitator-type transport system (GLF) for glucose and fructose (Parker et al. 1995; Weisser et al. 1995). Expression of glf, encoding the glucose facilitator protein from Z. mobilis from the low-copy plasmid pZY507glf in E. coli pET24[fdhmdh] allowed efficient transport of d-fructose into the cell without coupled phosphorylation (Parker et al. 1995; Weisser et al. 1995) and brought about a strong increase in the biotransformation efficiency [4.1 g·(g·h)–1] of the cells, which subsequently produced 362 mM d-mannitol under pH- control by formic-acid titration. A further increase in product concentration (500 mM) was achieved by pH-static and fed-batch addition of d-fructose to the biotransformation system.
Wild-type E. coli strains can grow with d-fructose in the growth medium at concentrations <2 mM. At these low concentrations, d-fructose is taken up and predominantly phosphorylated to fructose 1-phosphate via two membrane-associated proteins specified by fruA and fruB. At concentrations >2 mM, d-fructose is also taken up and phosphorylated (but to fructose 6-phosphate) via the membrane-associated uptake system for mannose, specified by manXYZ (Kornberg et al. 2000). These native PTS sugar-uptake systems of E. coli, delivering fructose 1-phosphate or fructose 6-phosphate into the cell, do not compete to an appreciable extent with the recombinant sugar facilitator. Whereas PTS is a high-affinity, low-velocity accumulative sugar transport system, GLF is a low-affinity, high-velocity concentration-dependent sugar facilitator (Parker et al. 1995; Weisser et al. 1995).
With respect to productivity and yield, whole-cell biotransformation with resting cells of E. coli pET24[fdhmdh] pZY507glf is as efficient as other production methods described so far, i.e. the utilization of Leuconostoc sp. resting cells in membrane cell-recycle cultures (von Weymarn et al. 2002), and a process employing purified mannitol dehydrogenase from Pseudomonas fluorescens and formate dehydrogenase from Candida boidinii (Slatner et al. 1998b) (Table 3). The three different biotechnological approaches lead to similar yields of 80–98 (mol%) and final concentrations (72–98 g/l) of d-mannitol. d-Mannitol production with lactic acid bacteria requires one-third of the fructose to be used in cofactor regeneration. This fraction is usually replaced by glucose so that the d-mannitol yield for the L. mesenteroides process refers to the initial fructose concentration and not to the total sugar content of the production medium (von Weymarn et al. 2002). However, a comparison of the two whole-cell biotransformation processes utilizing Leuconostoc spec. and E. coli shows that the specific d-mannitol productivity of the recombinant strain of E. coli is 1.8-fold higher than that of wild-type Leuconostoc species. Whereas both the recombinant E. coli process and the enzymatic synthesis produce d-mannitol with essentially no by-products, lactic acid and acetic acid are formed in the L. mesenteroides process. Furthermore, the cultivation of Leuconostoc species requires complex media. In the case of enzymatic synthesis, the biotransformation enzymes have to be isolated from recombinant E. coli cells (Slatner et al. 1998b).
By applying a recombinant E. coli strain overproducing the participating enzymes for d-mannitol production, the above-mentioned disadvantages of enzymatic synthesis and of Leuconostoc biotransformation can be successfully overcome by a whole-cell biotransformation system. This system makes use of the benefits of the two above-mentioned processes in implementing mannitol dehydrogenase from the natural producer of d-mannitol, L. pseudomesenteroides, and in establishing an in vivo recombinant enzymatic oxidation/reduction cycle.
References
Barnell WO, Yi KC, Conway T (1990) Sequence and genetic organization of a Zymomonas mobilis gene cluster that encodes several enzymes of glucose metabolism. J Bacteriol 172:7227–7240
Bradford MM (1976) A rapid and sensitive method for the quantitation of micrgram quantities of protein utilizing the principles of protein-dye binding. Anal Biochem 72:248–254
Bringer-Meyer S, Sahm H (1988) Acetoin and phenylacetylcarbinol formation by the pyruvate decarboxylases of Zymomonas mobilis and Saccharomyces carlsbergensis. Biocatalysis 1:321–331
Budesinsky Z, Protiva M (1961) Ephedrin. In: Knobloch W (ed) Synthetische Arzneimittel, Akademie-Verlag, Berlin, pp 24–27
Endo T, Koizumi S (2001) Microbial conversion with cofactor regeneration using genetically engineered bacteria. Adv Synth Catal 343:521–526
Galkin A, Kulakova L, Tishkov V, Esaki N, Soda K (1995) Cloning of formate dehydrogenase gene from a methanol-utilizing bacterium Mycobacterium vaccae N10. Appl Microbiol Biotechnol 44:479–483
Galkin A, Kulakova L, Yoshimura T, Soda K, Esaki N (1997) Synthesis of optically active amino acids from alpha-keto acids with Escherichia coli cells expressing heterologous genes. Appl Environ Microbiol 63:4651–4656
Hahn G, Kaup B, Bringer-Meyer S, Sahm H (2003) A zinc-containing mannitol-2-dehydrogenase from Leuconostoc pseudomesenteroides ATCC 12291: purification of the enzyme and cloning of the gene. Arch Microbiol 179:101–107
Haltrich D, Nidetzky B, Miemietz G, Gollhofer D, Lutz S, Stolz P, Kulbe KD (1996) Simultaneous enzymatic synthesis of mannitol and gluconic acid: I. Characterization of the enzyme system. Biocatal Biotrans 14:31–45
Hanahan D (1983) Studies on the transformation of E. coli with plasmids. J Mol Biol 166:557–580
Johnson JC (1976) Sugar alcohols and derivatives. In: Specialized sugars for the food industry. Noyes Data Corporation, NJ, p 313
Kataoka M, Rohani LPS, Yamamoto Y, Wada M, Kawabata H, Kita K, Yanase H (1997) Enzymatic production of ethyl(R)-4-chloro-3-hydroxybutanoate: asymmetric reduction of ethyl 4-chloro-3-oxobutanoate by an Escherichia coli transformant expressing the aldehyde reductase gene from yeast. Appl Microbiol Biotechnol 48:699–703
Kornberg HL (2001) Routes for fructose utilization by Escherichia coli. J Mol Microbiol Biotechnol 3:355–359
Kornberg HL, Lambourne LTM, Sproul AA (2000) Facilitated diffusion of fructose via the phosphoenolpyruvate/glucose phosphotransferase system of Escherichia coli. Proc Natl Acad Sci USA 97:1808–1812
Mahato SB, Garei S (1997). Advances in microbial steroid biotransformation. Steroids 62:332–345
Makkee M, Kieboom APG, van Bekkum H (1985) Production methods of d-mannitol. Starch/Stärke 37:136–141
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, pp 352–355
Murray HC, Petersen DH (1952). Oxygenation of steroids by Mucorales fungi. U. S. Patent 2602769 (Upjohn Co., Kalamazoo, Michigan, USA)
Nidetzky B, Haltrich D, Schmidt K, Schmidt H, Weber A, Kulbe KD (1996) Simultaneous enzymatic synthesis of mannitol and gluconic acid: II. Development of a continuous process for a coupled NAD(H)-dependent enzyme system. Biocatal Biotrans 14:47–65
Parker C, Barnell WO, Snoep JL, Ingram LO, Conway T (1995) Characterization of the Zymomonas mobilis glucose gacilitator gene product (glf) in recombinant Escherichia coli: examination of transport mechanism, kinetics and the role of glucokinase in glucose transport. Mol Microbiol 15:759–802
Reichstein T, Grüssner A (1934) Eine ergiebige Synthese der 1-Ascorbinsäure (C-Vitamin). Helv Chim Acta 17:311–328
Rogers PL, Shin HS, Wang B (1997). Biotransformation for l-ephedrine production. Adv Biochem Eng Biotechnol 56:33–59
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Schedel M (2000) Regioselective oxidation of aminosorbitol with Gluconobacter oxydans, key reaction in the industrial 1-deoxynojirimycin synthesis, p. 296–311. In: Rehm H-J, Reed G, Pühler A, Stadler P (eds) Biotechnology, vol 8b. Wiley-VCH, Weinheim
Schütte H, Flossdorf J, Sahm H, and Kula M-R (1976) Purification and properties of formaldehyde dehydrogenase and formate dehydrogenase from Candida boidinii. Eur J Biochem 62:151–160
Slatner M, Nagl G, Haltrich D, Kulbe KD, Nidetzky B (1998a) Enzymatic synthesis of mannitol. Reaction engineering for a recombinant mannitol dehydrogenase. Ann NY Acad Sci 864:450–453
Slatner M, Nagl G, Haltrich D, Kulbe KD, Nidetzky B (1998b) Enzymatic production of pure d-mannitol at high productivity. Biocatal Biotrans 16:351–363
Soetaert W, Buchholz K, Vandamme EJ (1995) Production of d-mannitol and d-lactic acid by fermentation with Leuconostoc mesenteroides. Agro-Food-Industry Hi-Tech 6:41–44
Soetaert W, Vanhooren PT, Vandamme EJ (1999) The production of mannitol by fermentation. In: Bucke C (ed) Methods in biotechnology, vol 10. Humana, Totowa New Jersey, pp 261–275
Von Weymarn N, Kiviharju K, Leisola M (2002) High-level production of d-mannitol with membrane cell-recycle bioreactor. J Ind Microbiol Biotechnol 29:44–49
Weisser P, Krämer R, Sahm H, Sprenger GA (1995) Functional expression of the glucose transporter of Zymomonas mobilis leads to restoration of glucose and fructose uptake in Escherichia coli mutants and provides evidence for its facilitator action. J Bacteriol 177:3351–3354
Acknowledgements
We wish to thank N. Esaki for plasmid pMcFDH, G.A. Sprenger for plasmid pZY507glf. We also thank the Fonds der Chemischen Industrie. This work was supported by the Institut für Technologie der Kohlenhydrate–Zuckerinstitut e. V.–Braunschweig, Germany. The authors ensure that all experiments comply with the current German laws.
Author information
Authors and Affiliations
Corresponding author
Additional information
A comment to this article is available at http://dx.doi.org/10.1007/s00253-015-7029-8.
Rights and permissions
About this article
Cite this article
Kaup, B., Bringer-Meyer, S. & Sahm, H. Metabolic engineering of Escherichia coli: construction of an efficient biocatalyst for d-mannitol formation in a whole-cell biotransformation. Appl Microbiol Biotechnol 64, 333–339 (2004). https://doi.org/10.1007/s00253-003-1470-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1470-9