
Abstract
The sipM gene of Bacillus megaterium encoding a type I signal peptidase (SPase) was isolated and structurally characterized. RNA analysis revealed a transcript size in accordance with a bicistronic operon comprising sipM and an adjacent open reading frame. Inactivation of sipM by targeted gene disruption could not be achieved, indicating its essential role for cell viability since there might be no other type I SPase of major importance present in B. megaterium. Plasmid-assisted amplification of the gene resulted in an increase in activity of the heterologous glucanase used as an extracellular reporter, suggesting a potential bottleneck for protein secretion within this species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Due to their high protein secretion capacity, members of the genus Bacillus are widely used for industrial production of extracellular enzymes (Debabov 1982; Priest 1989; Jarnagin and Ferrari 1992; Zukowski 1992; Ferrari et al. 1993). The majority of secreted proteins are transported across the cytoplasmic membrane via the Sec pathway; hence, they are initially synthesized as preproteins with a short amino-terminal extension, the signal peptide, which is required for targeting the precursor-protein to the secretion machinery located in the membrane (Fekkes and Driessen 1999). Removal of the signal peptide by signal peptidases (SPases) present on the extracytoplasmic face of the membrane is a prerequisite for the release of mature extracellular proteins from the membrane and is considered to be a bottleneck in the secretion pathway (Bolhuis et al. 1999).
Three types of SPases are known, of which type I is responsible for processing the majority of exported proteins; type II SPases exclusively process prelipoproteins, and type III enzymes specifically remove signal peptides of prepilin-like proteins (Tjalsma et al. 2000). A number of bacteria encode a multiple set of paralogous type I SPases. In Bacillus subtilis, seven genes for type I SPases have been identified (Bron et al. 1998): five of these (sipS, sipT, sipU, sipV, and sipW) are chromosomally located (Kunst et al. 1997), with the two additional genes (sipP1015 and sipP1040) being found on plasmids in natto-producing strains (Meijer et al. 1995). Two of the chromosomally encoded enzymes, SipS and SipT, are apparently of major significance as they have been shown to be essential for cell viability, i.e., creation of a double mutant was not possible (Tjalsma et al. 1998). Though all of the other type I SPases of B. subtilis are functional in processing preproteins, evidence from gene knock-out experiments points to their minor significance (Tjalsma et al. 1999). Recently, the presence of multiple, paralogous, type I SPases [SipS(ba), SipT(ba), SipV, and SipW] has been reported for Bacillus amyloliquefaciens (Chu et al. 2002). Preliminary sequence data of the Bacillus anthracis genome (http://www.tigr.org) also reveals at least six different loci coding for type I SPases. In marked contrast, Escherichia coli harbors only one type I SPase gene (lepB), which is essential for cell viability since a genetic knock-out could not be achieved (Date 1983). In Staphylococcus aureus, two potential type I SPase loci have been identified, but one of these (spsA) codes for a proteolytically inactive polypeptide; the active SPase I is encoded by the adjacent spsB gene, which again turned out to be essential (Cregg 1996).
Bacillus megaterium is a well-characterized and industrially important species (Vary 1992, 1993). It has gained considerable interest not only as a model for studying genetic systems in Gram-positive bacteria, such as regulation of the xylose-utilization operon (Rygus and Hillen 1992; Schmiedel et al. 1997), β-galactosidase expression (Strey et al. 1999; Shaw et al. 2002) and the induction of P450-cytochrome monooxygenases (Shaw and Fulco 1992), but also as a versatile producer of many valuable products, such as penicillin amidase and vitamin B12 (reviewed by Vary 1994), oxetanocin (Morita et al. 1999) and useful enzymes (Nagao et al. 1992; Wittchen and Meinhardt 1995). Due to the high stability of recombinant plasmids and low extracellular protease activity (Meinhardt et al. 1989; Von Tersch and Robbins 1990), it has also proven to be an efficient cloning host that appears to be especially attractive for production of extracellular enzymes. However, neither the amino acid sequence of a B. megaterium SPase nor the nucleotide sequence of the corresponding gene is available to date. Also, it is still unknown whether SPases constitute a bottleneck for expression of secreted proteins in this species.
Here, we report the isolation and characterization of sipM, a gene coding for a presumably essential type I SPase from B. megaterium DSM319. Functionality was proven by transcript analysis as well as plasmid-assisted co-expression of SipM and a heterologous glucanase used as an extracellular reporter, concomitantly resulting in an increase in glucanase activity.
Materials and methods
Bacterial strains and plasmids
The strains and plasmids used in this study are listed in Table 1.
Media and growth conditions
Bacteria were grown at 37°C in Luria-Bertani (LB) broth or in minimal medium (Sambrook et al. 1989) with 1% succinate or glucose as the carbon source, 0.1 mM CaCl2, 0.01% yeast extract, and 0.02% casamino acids. For antibiotic selection, Bacillus cultures were supplemented with tetracycline (12.5 μg/ml) and E. coli cultures with ampicillin (100 μg/ml). Screening for protease activity was performed on Ca-caseinate agar, prepared according to the manufacturer’s recommendations (Merck, Darmstadt, Germany). (1,3-1,4)-β-Glucanase activity was screened on LB plates containing 0.02% (w/v) lichenin and subsequent overlaying of grown colonies with an aqueous Congo red solution (0.2%, w/v).
Recombinant DNA techniques
Molecular cloning procedures were carried out as described in Sambrook et al. (1989). Plasmid DNA was purified using Jetstar columns (Genomed, Bad Oeynhausen, Germany). Preparation of genomic DNA from B. megaterium was performed as previously described (Gärtner et al. 1988). Reactions for in vitro amplification of DNA contained 200 μM dNTPs, 100 ng chromosomal template DNA, 100 pmol of each primer and 2.5 U Pwo DNA polymerase (PEQLab, Erlangen, Germany). Prior to amplification, the sample was heat-denatured for 3 min at 94°C and, after addition of the polymerase, 35 cycles were run. Isolation of restriction fragments or PCR products after gel electrophoresis was carried out with a Jetquick gel extraction kit (Genomed). Nucleotide sequences were determined with IRD81-labeled universal and reverse primers using the CycleReader Auto DNA sequencing kit (MBI Fermentas, St. Leon-Rot, Germany) and an automatic LI-COR sequencer (LI-COR, Lincoln, Neb.). For sequence assembly, analyses of the sequence and database searches, the HUSAR program package (EMBL, Heidelberg, Germany) and programs provided by the NCBI server (Bethesda, Md.) were used.
Hybridization procedures
For Northern analyses, total RNA from B. megaterium cultures grown under different conditions was isolated as previously described (Wittchen et al. 1998). Cells were harvested after 180 min (early exponential), 300 min (mid exponential), and 420 min (end exponential growth) from cultures grown in minimal medium with 1% succinate as the carbon source. An internal part (507 bp) of the sipM gene, synthesized by PCR as indicated in Fig. 1B applying primer pair sipM4 and sipM5 (5′-TGAACTGTGGGAATGGAT-3′ and 5′-GGCCAGTATACGGCACTC-3′), was labeled by in vitro transcription with a digoxigenin (DIG) RNA labeling kit (Roche, Mannheim, Germany). Following electrophoresis in 1.5% (w/v) formaldehyde agarose gels and transfer of nucleic acids to nylon membranes (Hybond-N, Amersham, Freiburg, Germany), hybridization was carried out at 45°C; detection of hybridizing bands was performed with a nucleic acid detection kit (Roche) using CSPD as the chemoluminescence substrate. Routinely, blotting membranes were exposed to X-ray films for 30–60 min (Hyperfilm-MP, Amersham).

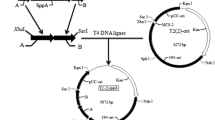
Genetic organization of the sipM-encoding genomic region of Bacillus megaterium DSM319. A Sequence of the intergenic region between rplS′ and sipM. Arrows putative transcriptional terminator; dotted line T-stretch; −35, −10 hypothetical σA-dependent sipM promoter elements; S/D ribosomal binding site. The protein sequence of coding regions is given in standard one-letter code beneath the nucleotide sequence. B Schematic representation of the sipM locus. Open reading frames (ORFs) are depicted as arrows, indicating transcriptional orientation. rplS′ truncated ORF upstream of sipM with similarities to the Bacillus subtilis 50S ribosomal protein L19, ORF1 ORF downstream of sipM largely homologous to the B. subtilis hypothetical protein YlqF, Hairpins potential Rho-independent transcriptional terminators. PCR primers M1, M2 and M4, M5 correspond to sipM1, sipM2 and sipM4, sipM5 in Materials and methods, respectively. The fragment amplified using M1 and M2, spanning the entire sipM gene, was used in expression studies; that obtained with M4 and M5 served as the template for labeling by in vitro transcription. C Sequence of the non-coding region downstream of ORF1. Arrows putative transcriptional terminator, dotted line T-stretch
Transformation of B. megaterium and single copy replacement
Protoplasts of B. megaterium were transformed by a PEG-mediated procedure (Vorobjeva et al. 1980; Meinhardt et al. 1989). Following transformation with temperature-sensitive plasmids, such as pUCTV2 and derivatives, regeneration of protoplasts was carried out on plates containing tetracycline (12.5 μg/ml) and incubated at the permissive temperature of 30°C. Curing of the plasmid was carried out by transferring single colonies to plates without the antibiotic and subsequent incubation at the non-permissive temperature of 42°C.
Amplification of sipM in B. megaterium and glucanase assay
A blunt-ended fragment (972 bp) comprising the entire sipM gene and its promoter region, synthesized by PCR (as indicated in Fig. 1B) with primers sipM1 (5′-TACGTGAATTACGTGGTAAAGCTTCGCG-3′) and sipM2 (5′-TTGGATTTCGCGAAGCTTGCGGAATGCG-3′), was inserted into the filled-in HindIII restriction site of the shuttle plasmid pBCmac, which carries the bgl gene (see Table 1). The resulting hybrid plasmid, pBCmac-sipM, was transformed into B. megaterium DSM319, whereas pBCmac lacking sipM was used for comparative analyses. Activity of the (1,3-1,4)-β-glucanase, encoded by the bgl gene of Paenibacillus macerans (Borriss et al. 1990), was monitored by measuring the amount of reducing sugar released due to enzymatic hydrolysis of lichenin (Miller 1959). Along with wild-type B. megaterium DSM319, which served as the control, transformants were cultivated in minimal medium with 1% succinate as the carbon source and 12.5 μg/ml tetracycline. Growth was monitored by measuring the optical density (OD546) during cultivation. After 300 min, samples for enzyme assays were collected by centrifugation at hourly intervals. Reaction mixtures, containing 180 μl distilled water, 30 μl 0.5 M Tris-base buffer (adjusted to pH 7.2 with HCl), 150 μl lichenin solution (5 mg/ml), and 75 μl supernatant, were incubated at 30°C for 10 min. Reactions were terminated by addition of 450 μl 3,5-dinitrosalicylic acid (DNS) solution (1.6% NaOH, 1% DNS, 30% Rochelle salt) and boiling for 10 min. Finally, absorption was measured at 547 nm. Miller units (Miller 1972) were calculated by relating the volume activity (nkat/ml) to the corresponding OD of the culture.
Nucleotide sequence accession number
The 2,200-bp chromosomal region of B. megaterium DSM319 carrying the entire sipM gene, a partial rplS′ gene (upstream of sipM) and the complete ORF1 (immediately downstream of sipM) was sequenced on both strands and has been submitted to the EMBL database under accession no. AJ549327.
Results
Isolation, cloning and sequencing of the sipM locus
We previously constructed a B. megaterium DSM319 mutant (MS941) that lacks 98.5% of its extracellular protease activity due to the knock-out of the nprM gene, which encodes a neutral metalloprotease (Wittchen and Meinhardt 1995). During the search for additional peptidases in a plasmid gene library of mutant MS941, we obtained a candidate clone that displayed faint proteolytic activity on Ca-caseinate agar (not shown); it contained a recombinant plasmid designated as pUCKM1 (see Table 1). Restriction analysis revealed the insert to be a fragment of 4.5 kb, from which a 2.2-kb fragment was subcloned and characterized by sequencing on both strands. A complete open reading frame (ORF) homologous to SPase I genes of Gram-positive bacteria was identified on this fragment; accordingly, the gene was designated sipM (Fig. 1B). In NCBI-BLAST searches (Altschul et al. 1997), the predicted polypeptide revealed highest identities to enzymes of B. anthracis (54%), Bacillus caldolyticus (52%), and Listeria monocytogenes (50%). An alignment of type I SPases with high similarity scores to SipM of B. megaterium DSM319 is presented in Fig. 2; the polypeptide contains residues known to be crucial conformational determinants (R81, D146), but also residues essential for enzymatic activity (S39, K80) (van Dijl et al. 1995; Paetzel et al. 2002). The coding region of sipM spans 552 bp; hence, the predicted protein consists of 183 amino acid residues with a calculated molecular mass of 20.5 kDa. There is a potential ribosome binding site (GGAGG) seven nucleotides upstream of the start codon (ATG) and a typical Bacillus promoter sequence motif is situated further upstream (Fig. 1A). Immediately downstream of sipM, a second complete ORF (ORF1) with the same transcriptional direction was identified; the first two-thirds of the predicted polypeptide exhibits 74% identity to B. subtilis YlqF, a hypothetical protein displaying similarities to GTPases. A putative Rho-independent terminator structure can be found eight nucleotides downstream of the ORF1 stop codon (see Fig. 1C). Upstream of sipM a truncated ORF was sequenced which exhibits striking similarities (89% identity) to the B. subtilis 50S ribosomal protein L19; thus, it was designated rplS′.

Amino acid sequence alignments of five type I signal peptidases (SPases) selected from an NCBI-BLAST list of best hits to the predicted SipM protein of B. megaterium DSM319. Residues essential for the activity or stability of the protein are marked by triangles. Bme B. megaterium DSM319-SipM, Ban Bacillus anthracis SPase I (NP_655047), Bca Bacillus caldolyticus SPase I (Swissprot: P41027), Lmo Listeria monocytogenes SPase I (Sptrembl: Q8Y7K6), Sau Staphylococcus aureus SPase IB (Swissprot: P72365), Bsu B. subtilis SPase S (Swissprot: P28628)
Transcript analysis of the sipM locus
Since nothing was known about expression of sipM, we checked its transcription during different growth phases in culture. For this purpose, B. megaterium DSM319 was cultivated in minimal medium with 1% succinate as the carbon source and cells were harvested at early, mid, and late exponential growth (see Materials and methods for details). Equal amounts of total RNA were subjected to gel electrophoresis and analyzed by Northern blotting. An in vitro transcribed DIG-labeled RNA prepared from a DNA fragment amplified by PCR using primer pair M4 and M5 (see Fig. 1B) served as the probe. As depicted in Fig. 3, the sipM transcript was detected in all cases. The strongest signal was obtained during mid exponential growth (Fig. 3, lane B). The size of transcript estimated from its electrophoretic mobility is approximately 1.8 kb, which is a reasonable match to the postulated mRNA length of a bicistronic operon comprising sipM and ORF1 as indicated in Fig. 1B.

Northern analysis of sipM expression. Equal amounts of total RNA isolated from DSM319 cultures grown in minimal medium containing 1% succinate as the carbon source at different growth phases were separated in RNA gels and transferred to nylon membranes. Hybridization was performed at 45°C using an in vitro transcribed digoxygenin (DIG)-labeled RNA sipM probe. Lanes: A early exponential growth, B mid exponential growth, C late exponential growth
Gene knock-out experiments
As for other Bacilli, targeted gene disruption in B. megaterium is a two-step process that initially needs recombinative integration of the entire vector carrying the disruption cassette. A second recombination event leads to excision of the vector either via the same flank, thereby restoring the wild-type situation in the chromosome, or via the other flank, eventually leading to exchange of the chromosomal gene copy for the disrupted one. Subsequent curing of the vector establishes a stable mutant genotype (Rygus and Hillen 1992). Inactivation of the sipM locus in B. megaterium DSM319 was attempted by applying a previously developed and for a number of loci successfully used temperature-sensitive, single-copy replacement vector (Wittchen and Meinhardt 1995; Wittchen et al. 1998; Strey et al. 1999; Lee et al. 2001). The constructed hybrid plasmid, pDSIP2, carries a disrupted copy of sipM created by integrating the bgl gene of P. macerans, encoding an extracellular (1,3-1,4)-β-glucanase, into its single HindIII site (Borriss et al. 1990). Following PEG-mediated transformation of B. megaterium DSM319, tetracycline-resistant clones were selected and treated as described in Materials and methods to eliminate the plasmid. All attempts to isolate a clone that had lost its tetracycline resistance (i.e., the vector), but still retained the bgl gene, failed. Clones analyzed in detail after the curing procedure had either lost the plasmid without allele exchange or pDSIP2 was still integrated in the chromosome (data not shown); in either case there is an intact copy of sipM.
Functional expression studies on sipM
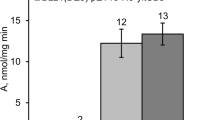
Proteolytic processing of preproteins carrying signal peptides is a limiting step for protein secretion in that it constitutes a bottleneck for the export of extracellular enzymes (van Dijl et al. 1991; Bolhuis et al. 1999). To gain further evidence for the functionality of the sipM gene product, co-expression of sipM and bgl (encoding the β-glucanase of P. macerans—already used in the gene knock-out experiments) was performed by placing both genes on a joint replicon. A 927 bp PCR-generated fragment (see primers M1 and M2 in Fig. 1B) comprising the entire sipM gene (coding region as well as expression signals) was integrated into the E. coli/Bacillus shuttle plasmid pBCmac carrying the bgl gene. Since B. megaterium does not encode a (1,3-1,4)-β-glucan-hydrolyzing enzyme itself (Wittchen et al. 1998), enzymatic activities of transformants harboring either pBCmac or the hybrid plasmid pBCmac-sipM could easily be detected (Fig. 4). As to be expected, no β-glucanase activity was detected in B. megaterium DSM319, whereas in the culture supernatants of both plasmid-carrying strains, enzyme activities accumulated with cultivation time. However, at each of the five time points, the pBCmac-sipM-containing culture displayed a clear increase (at least 44%) in extracellular glucanase activity when compared to the pBCmac-harboring strain.

β-Glucanase activities of B. megaterium DSM319 wild-type, DSM319 (pBCmac) and DSM319 (pBCmac-sipM). Strains were cultivated in minimal medium containing 1% succinate. After 300 min of cultivation, samples were taken at hourly intervals. β-Glucanase activity was determined by measuring the amount of reducing sugar released due to hydrolysis of lichenin. Miller units were calculated by relating the volume activities to the corresponding OD at 546 nm. The values given are the means of two independent experiments. Glucanase activity is observed only in plasmid-carrying strains (pBCmac and pBCmac-sipM). Co-expression of the P. macerans bgl (β-glucanase) reporter gene with sipM results in an increase in activity of at least 40% at all time points
Discussion
In the course of screening for peptidases in B. megaterium mutant strain MS941 displaying only 1.5% of wild-type extracellular protease activity (Wittchen and Meinhardt 1995), we isolated a locus encoding a type I SPase (sipM) from a plasmid gene library. The corresponding E. coli clone harbored a 4.5 kb chromosomal insert and caused hardly detectable haloes on Ca-caseinate agar. This faint proteolytic activity could be ascribed to a segment of 2,200 bp. Sequencing revealed one truncated and two complete ORFs, one of which was identified as a gene encoding a peptidolytic protein, namely sipM. Although we identified sipM-carrying E. coli clones because of their peptidolytic activities, we do not consider such a procedure to be a generally applicable method for the screening of SPases, since it cannot be decided whether the observed phenotype is due to heterologous sipM expression or if it is simply coincidental.
The size of the sipM gene is in good accordance with other type I SPases genes of Gram-positive bacteria. Also, the predicted protein sequence reveals significant homologies to enzymes of B. anthracis, B. caldolyticus, L. monocytogenes, S. aureus, and B. subtilis. Amino acid residues considered to be essential for stability and activity of bacterial type I SPases are conserved (for details see Fig. 2). Recent investigations on the enzymology of type I SPases favor a mechanism based on a catalytic dyad consisting of serine and lysine rather than the previously assumed catalytic triad (Black 1993; Paetzel et al. 2002). Thus, SipM and its homologues belong to a newly arranged class of serine proteases, like those of the LexA-like proteins (van Dijl et al. 1995; Peng et al. 2001).
The potential ribosome binding site (S/D) located seven nucleotides in front of the ATG start codon matches perfectly the Bacillus consensus sequence GGAGG (Vellanoweth and Rabinowitz 1992). The putative promoter motif situated further upstream, with −10- and −35- elements similar to vegetative σA-dependent promoters of B. subtilis (Moran et al. 1982), is consistent with transcription occurring during the exponential growth phase; its location is also in agreement with the size of the transcript. Immediately downstream of sipM, only 21 nucleotides away from its stop codon, a second complete ORF (ORF1) with the same transcriptional orientation is present. The protein deduced from the 1,086 bp long coding region reveals that the first two-thirds are homologous to the ylqF gene product of B. subtilis (74% identity), a hypothetical protein (846 bp; SubtiList database: BG13405) with similarities to GTPases. Located upstream of sipM is a truncated coding region of 300 bp (rplS′) encoding a polypeptide with striking similarities (89% identity) to the 50S ribosomal protein of B. subtilis L19 (354 bp; SubtiList database: BG12667). Unlike in B. subtilis, where the rplS gene is adjacent to the monocistronically organized yqlF, sipM of B. megaterium is situated in between these two genes, presumably indicative of quite a large evolutionary distance between these two species. Since transcriptional terminator structures (one succeeding rplS′ and another downstream of ORF1; see Fig. 1 for details) and no promoter elements upstream of ORF1 could be identified, the locus comprising sipM and ORF1 appears to be bicistronically organized with an estimated transcript length of 1.8 kb. The size of the transcript was confirmed when total RNA isolated from cells at different growth phases (early, mid, and late exponential growth) were subjected to Northern analysis. Hybridizing bands with an electrophoretic mobility corresponding to the expected size were detected in all three instances (Fig. 3). The strongest signals were routinely obtained in samples taken during mid exponential growth phase. Expression patterns as depicted in Fig. 3 were also obtained in cultures grown in 1% glucose instead of succinate as well as in LB broth (data not shown). However, growth phase-dependent regulation of sipM expression cannot be concluded, because that would require an internal standard for exact transcript quantification.
Studying functionality of a gene and the encoded protein is often performed by gene knock out experiments. Therefore, an in vitro-inactivated copy of sipM was placed on the temperature-sensitive, single-copy replacement vector pUCTV2 (Wittchen and Meinhardt 1995) and transformed into the wild-type strain B. megaterium DSM319. SipM is apparently not dispensable for cell viability, since all attempts to isolate a mutant strain after the curing procedure failed. As in other bacteria, if there is only one major type I SPase, the corresponding gene cannot be knocked out without lethal consequences (Date 1983; Cregg 1996). Consistent with such findings, we did not detect any other sipM-paralogous genes in B. megaterium DSM319 by Southern hybridization studies under low-stringent conditions using sipM as a probe (data not shown). Thus, sipM presumably represents the only locus encoding a type I SPase of major significance in B. megaterium DSM319. However, the existence of additional type I SPases of minor importance cannot be ruled out, and indeed sipW-like genes have recently been identified in a number of Bacilli, amongst them B. megaterium (Chu et al. 2002).
Since targeted gene disruption as a means of proving functionality of sipM was not possible, co-expression of the signal peptidase and the (1,3-1,4)-β-glucanase of P. macerans as an extracellular reporter was performed. Such transformants displayed the highest values of glucanase activity with an increase of not less than 40% at each reading point when compared to clones lacking the plasmid-based sipM. Such a clear increase in extracellular enzyme activity upon amplification of the gene proves not only functionality of sipM, but also its ability to enhance translocation efficiency, presumably by opening a potential bottleneck for protein secretion in B. megaterium.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Black MT (1993) Evidence that the catalytic activity of prokaryote leader peptidase depends upon the operation of a serine-lysine catalytic dyad. J Bacteriol 175:4957–4961
Bolhuis A, Tjalsma H, Smith HE, de Jong A, Meima R, Venema G, Bron S, van Dijl JM (1999) Evaluation of bottlenecks in the late stages of protein secretion in Bacillus subtilis. Appl Environ Microbiol 65:2934–2941
Borriss R, Buettner K, Maentsaelae P (1990) Structure of the beta-1,3-1,4-glucanase gene of Paenibacillus macerans: homologies to other beta-glucanases. Mol Gen Genet 222:278–283
Bron S, Bolhuis A, Tjalsma H, Holsappel S, Venema G, van Dijl JM (1998) Protein secretion and possible roles for multiple signal peptidases for precursor processing in Bacilli. J Biotechnol 64:3–13
Chu HH, Hoang V, Kreutzmann P, Hofemeister B, Melzer M, Hofemeister J (2002) Identification and properties of type I-signal peptidases of Bacillus amyloliquefaciens. Eur J Biochem 269:458–469
Cregg KM (1996) Molecular cloning and expression of the spsB gene encoding an essential type I signal peptidase from Staphylococcus aureus. J Bacteriol 178:5712–5718
Date T (1983) Demonstration by a novel genetic technique that leader peptidase is an essential enzyme of Escherichia coli. J Bacteriol 154:76–83
Debabov VG (1982) The industrial use of Bacilli. In: Dubnau DA (ed) The molecular biology of the Bacilli, vol 1. Academic Press, New York, pp 331–370
Fekkes P, Driessen AJ (1999) Protein targeting to the bacterial cytoplasmic membrane. Microbiol Mol Biol Rev 63:161–173
Ferrari E, Jarnagin AS, Schmidt BF (1993) Commercial production of extracellular enzymes. In: Sonenshein AL, Hoch JA, Losick R (eds) Bacillus subtilis and other Gram-positive bacteria. American Society for Microbiology, Washington, D.C., pp 917–937
Gärtner D, Geissendörfer M, Hillen W (1988) Expression of Bacillus subtilis xyl operon is repressed at the level of transcription and is induced by xylose. J Bacteriol 170:3102–3109
Jarnagin AS, Ferrari E (1992) Extracellular enzymes: gene regulation and structure function relationship studies. In: Doi RH, McGloughlin M (eds) Biology of Bacilli: applications to industry. Butterworth-Heinemann, Stoneham, Mass., pp 191–219
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S et al (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Lee JS, Wittchen KD, Stahl C, Strey J, Meinhardt F (2001) Cloning, expression, and carbon catabolite repression of the bamM gene encoding beta-amylase of Bacillus megaterium DSM319. Appl Microbiol Biotechnol 56:205–211
Meijer WJ, de Jong A, Bea G, Wisman A, Tjalsma H, Venema G, Bron S, van Dijl JM (1995) The endogenous Bacillus subtilis (natto) plasmids pTA1015 and pTA1040 contain signal peptidase-encoding genes: identification of a new structural module on cryptic plasmids. Mol Microbiol 17:621–631
Meinhardt F, Stahl U, Ebeling W (1989) Highly efficient expression of homologous and heterologous genes in Bacillus megaterium. Appl Microbiol Biotechnol 41:344–351
Miller GL (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Miller JH (1972) Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp 352–355
Moran CP Jr, Lang N, LeGrice SF, Lee G, Stephens M, Sonenshein AL, Pero J, Losick R (1982) Nucleotide sequences that signal the initiation of transcription and translation in Bacillus subtilis. Mol Gen Genet 186:339–346
Morita M, Tomita K, Ishizawa M, Takagi K, Kawamura J, Takahashi H, Morino T (1999) Cloning of oxetanocin A biosynthetic and resistance genes that reside on a plasmid of Bacillus megaterium strain NK84-0128. Biosci Biotechnol Biochem 63:563–566
Nagao T, Mitamura T, Wang XH, Negoro S, Yomo T, Urabe I, Okada H (1992) Cloning, nucleotide sequences, and enzymatic properties of glucose dehydrogenase isozymes from Bacillus megaterium IAM1030. J Bacteriol 174:5013–5020
Paetzel M, Dalbey RE, Strynadka NC (2002) Crystal structure of a bacterial signal peptidase apoenzyme: implications for signal peptide binding and the Ser-Lys dyad mechanism. J Biol Chem 277:9512–9519
Peng SB, Wang L, Moomaw J, Peery RB, Sun PM, Johnson RB, Lu J, Treadway P, Skatrud PL, Wang QM (2001) Biochemical characterization of signal peptidase I from Gram-positive Streptococcus pneumoniae. J Bacteriol 183:621–627
Priest FG (1989) Products from Bacilli. In: Harwood CF (ed) Handbooks of biotechnology, vol 2. Plenum, New York, pp 293–315
Rygus T, Hillen W (1992) Catabolite repression of the xyl operon in Bacillus megaterium. J Bacteriol 174:3049–3055
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Schmiedel D, Kintrup M, Kuster E, Hillen W (1997) Regulation of expression, genetic organization and substrate specificity of xylose uptake in Bacillus megaterium. Mol Microbiol 23:1053–1062
Shaw GC, Fulco AJ (1992) Barbiturate-mediated regulation of expression of the cytochrome P450BM-3 gene of Bacillus megaterium by Bm3R1 protein. J Biol Chem 267:5515–5526
Shaw GC, Chiou CY, Chou YH, Li JM (2002) mbgA-dependent lactose utilization by Bacillus megaterium. Curr Microbiol 44:102–105
Stahl U, Esser K (1983) Plasmid heterogeneity in various strains of Bacillus megaterium. Eur J Appl Biotechnol 17:248–251
Strey J, Wittchen KD, Meinhardt F (1999) Regulation of beta-galactosidase expression in Bacillus megaterium DSM319 by a XylS/AraC-type transcriptional activator. J Bacteriol 181:3288–3292
Tjalsma H, Bolhuis A, van Roosmalen ML, Wiegert T, Schumann W, Broekhuizen CP, Quax WJ, Venema G, Bron S, van Dijl JM (1998) Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev 12:2318–2331
Tjalsma H, van den Dolder J, Meijer WJ, Venema G, Bron S, van Dijl JM (1999) The plasmid-encoded signal peptidase SipP can functionally replace the major signal peptidases SipS and SipT of Bacillus subtilis. J Bacteriol 181:2448–2454
Tjalsma H, Bolhuis A, Jongbloed JD, Bron S, van Dijl JM (2000) Signal peptide-dependent protein transport in Bacillus subtilis: a genome-based survey of the secretome. Microbiol Mol Biol Rev 64:515–547
Van Dijl JM, de Jong A, Smith H, Bron S, Venema G (1991) Signal peptidase I overproduction results in increased efficiencies of export and maturation of hybrid secretory proteins in Escherichia coli. Mol Gen Genet 227:40–48
Van Dijl JM, de Jong A, Venema G, Bron S (1995) Identification of the potential active site of the signal peptidase SipS of Bacillus subtilis. Structural and functional similarities with LexA-like proteases. J Biol Chem 270:3611–3618
Vary PS (1992) Development of genetic engineering in Bacillus megaterium: an example of the versatility and potential of industrially important Bacilli. In: Doi RH, McGloughlin M (eds) Biology of Bacilli: applications to industry. Butterworth-Heinemann, Stoneham, Mass., pp 253–310
Vary PS (1993) The genetic map of Bacillus megaterium. In: Ganesan AT, Hoch JA (eds), Genetics and biotechnology of Bacilli, vol 2. Academic Press, New York, pp 475–481
Vary PS (1994) Prime time for Bacillus megaterium. Microbiology 140:1001–1013
Vellanoweth RL, Rabinowitz JC (1992) The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol 6:1105–1114
Von Tersch MA, Robbins HL (1990) Efficient cloning in Bacillus megaterium: comparison to Bacillus subtilis and Escherichia coli cloning hosts. FEMS Microbiol Lett 58:305–309
Vorobjeva I, Khemel A, Alföldi I (1980) Transformation of Bacillus megaterium protoplasts by plasmid DNA. FEMS Microbiol Lett 7:261–263
Wittchen KD, Meinhardt F (1995) Inactivation of the major extracellular protease from Bacillus megaterium DSM319 by gene replacement. Appl Microbiol Biotechnol 42:871–877
Wittchen KD, Strey J, Bultmann A (1998) Molecular characterization of the operon comprising the spoIV gene of Bacillus megaterium DSM319 and generation of a deletion mutant. J Gen Appl Microbiol 44:317–326
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119
Zukowski MM (1992) Production of commercially valuable products. In: Doi RH, McGloughlin M (eds) Biology of Bacilli: applications to industry. Butterworth-Heinemann, Stoneham, Mass., pp 311–337
Acknowledgements
This work was supported by grants from the Federal Ministry of Education and Research (BMBF, Bonn-Bad Godesberg, Germany), grant no. 0319179E and 0312613. M.A. Rachman thanks the German Academic Exchange Service (DAAD, Bonn-Bad Godesberg, Germany) for financial support. The authors wish to thank K. Müller (Münster, Germany) for cloning the fragment carrying sipM in partially fulfillment of her diploma thesis, and J. Paluszynski (Münster, Germany) for checking the language.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nahrstedt, H., Wittchen, K.D., Rachman, M.A. et al. Identification and functional characterization of a type I signal peptidase gene of Bacillus megaterium DSM319. Appl Microbiol Biotechnol 64, 243–249 (2004). https://doi.org/10.1007/s00253-003-1469-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1469-2