
Abstract
We studied the growth characteristics and oxidative capacities of Acetobacter aceti IFO 3281 in batch and chemostat cultures. In batch culture, glycerol was the best growth substrate and growth on ethanol occurred only after 6 days delay, although ethanol was rapidly oxidized to acetic acid. In continuous culture, both glycerol and ethanol were good growth substrates with similar characteristics. Resting cells in a bioreactor oxidized ribitol to l-ribulose with a maximal specific rate of 1.2 g g−1 h−1). The oxidation of ribitol was inhibited by ethanol but not by glycerol. Biomass yield (YSX; C-mmol/C-mmol) on ethanol and glycerol was low (0.21 and 0.17, respectively). In the presence of ribitol the yield was somewhat higher (0.25) with ethanol but lower (0.13) with glycerol, with respectively lower and higher CO2 production. In chemostat cultures the oxidation rate of ribitol was unaffected by ethanol or glycerol. Cell-free extract oxidized ethanol very slowly but not ribitol; the oxidative activity was located in the cell membrane fraction. Enzymatic activities of some key metabolic enzymes were determined from steady-state chemostat with ethanol, glycerol, or ethanol/glycerol mixture as a growth limiting substrate. Based on the measured enzyme activities, metabolic pathways are proposed for ethanol and glycerol metabolism.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Acetic acid bacteria are well known for their ability to convert ethanol to acetic acid. Although the strains in this family have been studied comprehensively and used in industrial processes, only a few metabolic studies are available. According to Nakayama and De Ley (1965), acetic acid bacteria have two enzymatic systems for the conversion of ethanol to acetaldehyde: one is linked to the cytoplasmic membrane and the other is in the cytoplasm. The membrane-bound alcohol dehydrogenase (ADH), first reported by Nakayama (1961b), was later characterized by Matsuhita et al. (1992) as being a quinohemoprotein–cytochrome c complex located on the periplasmic side of the cytoplasmic membrane. According to Nakayama (1961b), the membrane-bound ADH has more than a hundred times higher specific activity than the soluble enzyme. Likewise, acetaldehyde dehydrogenase is present both as a membrane-bound cofactor-independent enzyme (Nakayama 1961a) and a cofactor-dependent soluble enzyme (Nakayama and De Ley 1965).
Reichstein (Reichstein and Grussner 1934) was the first to notice bioconversion of ribitol to ribulose by an unidentified acetic acid bacterium. Bhuiyan et al. (1998) studied this phenomenon later in the production of l-lyxose from ribitol in a two-step process involving acetic acid bacteria and immobilized l-rhamnose isomerase and d-tagatose 3-epimerase. Recently, Adachi et al. (2001) showed that ribitol oxidation is carried out by two different enzymes; one NAD(P)-dependent cytosolic enzyme and another membrane-bound non-NAD(P)-dependent enzyme. The cytosolic enzyme oxidized ribitol to d-ribulose and the membrane-bound to l-ribulose. Interest in the production of rare l-pentose sugars such as l-xylose and l-ribose is rapidly growing owing to their use in the manufacture of antiviral drugs (Scott 2002).
We have studied the production of l-ribose and l-arabinose from ribitol which is oxidized to l-ribulose by acetic acid bacteria and converted to l-ribose and l-arabinose by the recently discovered epimerase activity of xylose isomerase (Pastinen et al. 1999), using cross-linked enzymes in a column reactor (Leisola et al. 2001; Jokela et al. 2002). In this study we characterized the growth and ribitol oxidation capacities of Acetobacter aceti IFO 3281 in batch and chemostat cultures. We also studied some key enzyme activities in metabolism of glycerol and ethanol and suggest metabolic pathways for these substrates.
Materials and methods
Organisms, maintenance, and inocula preparation
Acetobacter aceti IFO 3281 was stored as a frozen stock culture containing 10% (v/v) glycerol in 2 ml ampoules at −80°C. The inoculum for shake flask cultivations was prepared by cultivating A. aceti in a 250 ml Erlenmeyer flask containing 50 ml of the complex medium at 30°C for 20–24 h.
For bioreactor cultivations, and for bioconversion experiments in shake flasks and in a bioreactor, the cells from the preculture were separated by centrifugation (8,000 rpm, 10 min) and then suspended in 50 ml of deionized water to give an initial biomass concentration of 0.1–0.3 g/l cell dry weight (CDW) in the bioreactor. For bioconversion reactions, the cells were suspended in 50 mM sodium phosphate or maleate buffer, pH 7, to give an initial CDW of 2.6–3.0 g/l in shake flasks and 1.7 g/l in the bioreactor.
For bioconversion experiments in a Bioscreen C automatic incubator, the cells were cultivated in a complex medium having 1% (w/v) glycerol as a carbon source. The cells for the experiment were separated (8,000 rpm, 10 min) and resuspended in 50 mM sodium phosphate or maleate buffer, pH 7.
Media
The complex medium contained yeast extract (Difco Laboratories, USA), 10 g/l; trypticase peptone (Becton Dickinson, USA), 10 g/l; KH2PO4,0.125 g/l; MgSO4·7 H2O, 0.25 g/l; and glycerol (Becton Dickinson) 10 g/l.
The mineral medium was prepared according to Verduyn (1991) and contained NH4(SO4)2, 5.0 g /l; KH2PO4,3.0 g/l; MgSO4·7 H2O, 0.5 g/l; EDTA, 15.0 mg/l; ZnSO4·7 H2O, 4.5 mg/l; CoCl2·6 H2O, 0.3 g/l; MnCl2·4 H2O, 1.03 g/l; CuSO4·5 H2O, 0.3 g/l; CaCl2·2 H2O, 4.5 g/l; FeSO4·7 H2O, 3.0 g/l; NaMoO4·2 H2O, 0.4 g/l; H3BO4, 1.0 mg/l; KI, 0.1 mg/l; and silicone antifoaming agent, 0.05 mg/l (BDH Laboratory Supplies, UK). After sterilization, a filter-sterilized (Sterivex-GV, 0.22 μm; Millipore, USA) vitamin solution was added, giving a final concentration of biotin 1.05 mg/l, calcium panthotenate 1.0 mg/l, nicotinic acid 1.5 mg/l, myo-inositol 25 mg/l, pyridoxal hydrochloride 1.5 mg/l, para-aminobenzoic acid 1.0 mg/l, riboflavin 1.5 mg/l, folic acid 1.0 mg/l, and vitamin B12 1.0 mg/l.
In the chemostat cultivations, three carbon sources and their (1+1) combinations (C-mmol/C-mmol) were investigated. Ethanol was added to the medium without sterilization. Glycerol was heat sterilized and ribitol (Pfanstiehl, USA) was filter-sterilized separately before addition. The pH of the medium was adjusted to 5.8 in all the growth experiments.
Cultivation conditions
Shake flask cultivations were done in Erlenmeyer flasks (250 ml, 200 rpm) containing 50 ml of the mineral medium at 30°C for 48–52 h. The medium contained 5–10 g/l of the various substrates and their combinations.
Batch and chemostat cultivations were carried out in a 3 l bioreactor (Biostat MD; B. Braun Melsungen, Germany) with a working volume of 1.5 l and automatic pH and temperature control. The cultivation temperature was 30°C and stirrer speed 600 rpm. The culture pH was kept at 5.8 by the addition of 3 M KOH and 0.5 M H2SO4. The airflow rate was set to maintain the dissolved oxygen concentration above 30% and it was controlled with a mass flow controller (Bronkhurst HiTech, Ruurlo, The Netherlands).
In the chemostat cultivations, the working volume was kept constant by removing the effluent with a peristaltic pump (Watson-Marlow 101U, UK) connected to a PID-controlled level probe. The dry weight of the cultivation, O2 consumption, and CO2 production were measured on successive days to confirm that a steady state had been established.
Bioconversion experiments
Bioconversion experiments were carried out at 30°C in 50 mM maleate buffer (pH 7). First experiments were done in Bioscreen C (Labsystems Oy, Finland) automatic incubator, which allows 200 samples to be run at a time in the 500-μl wells of two honeycomb plates. 350 μl of the cell suspension in a buffer solution was added to 50 μl of substrate dissolved in the corresponding buffer. The wells were incubated for 3 days with continuous shaking using 100 g/l ribitol, xylitol or l-arabitol as substrate.
Bioconversion experiments in shake flasks (200 rpm) were carried out with 50 g/l ribitol for 4 days.
Bioconversion experiments in 1-liter bioreactors (Biostat Q; Braun Biotech International, Germany) were carried out with a working volume of 500 ml at 800 rpm. Aeration was set at 1 vvm. The cells were incubated with 50 g/l ribitol for 21 h.
Analytical methods
Gas analysis
Carbon dioxide, oxygen, argon, and nitrogen were determined by mass spectrometer (VG-Prima 600, UK). For calculation of the rates of oxygen consumption and carbon dioxide production a temperature of 30°C was assumed and air pressure was taken from the daily weather report.
Optical density
The optical density of the cell suspension at 600 nm was measured using a spectrophotometer (Shimadzu UV-2100, Japan). The samples were diluted to obtain an optical density in the range 0.1–1.0.
Dry weight measurement
Culture samples (10 ml) were vacuum-filtered through a pre-dried and weighed nitrocellulose filter (0.45 μm; Schleicher and Schuell, Germany), washed with Milli-Q water, and dried in a microwave oven for 20 min (Ignis, Japan). Two parallel measurements were made on each sample.
Substrate and metabolite analysis
Ethanol, glycerol, acetate, succinate, and lactate concentrations were determined by HPLC using an HPX-87H+ Aminex ion-exclusion column (300×7.8 mm; Bio-Rad, USA) and detected with a Waters (USA) 410 refractive index detector and a Waters 486 UV detector. The column was eluted at 65°C with 5 mM H2SO4 at a flow rate of 0.6 ml/min. Ribitol, ribulose, and xylitol were separated on an HPX-87P Aminex ion-exclusion column (300×7.8 mm; BioRad) and detected with a Shimadzu (Japan) refractive index detector. The column was eluted at 85°C with H2O at a flow rate of 0.6 ml/min. The standard error of the analytical methods was less than 5%, except for ribulose. The ribulose quantification gave results that were too high compared with the theoretical yields. This is due to the lability of ribulose in the standard and in the sample. Therefore ribulose concentrations were estimated based on ribitol consumed. No other sugar metabolites were detected in ribitol bioconversion experiments.
Carbon balance and degree of reduction balance
An estimation of 50% carbon content of E. coli cells (Neidhardt et al. 1990) was used in calculations of the elemental carbon content of the cell dry weight samples. The carbon balance was calculated as the ratio of carbon recovery in metabolic products, including biomass, to the consumption of substrate carbon. The degree of reduction balance was calculated on the basis of metabolites produced and oxygen consumed.
Preparation of cell-free extract
Cells were harvested by centrifugation at +4°C, washed once with 10 mM potassium phosphate buffer (pH 7.5, +4°C) and resuspended in 4 ml sonication buffer (100 mM potassium phosphate, 2 mM MgCl2, 1 mM DDT, pH 7.5). Cells were disrupted by sonication with 0.7 mm glass beads at 0°C in a Vibra Cell sonicator (VCX600, USA). Ten intervals of 30 s pulses were applied with a 30 s cooling period between each pulse. Whole cells and cell debris were removed by centrifugation at +4°C and the clear supernatant, containing typically 2–5 mg protein/ml, was used for enzyme assays.
Protein content
Protein content of the whole cell samples was determined by a modified biuret method (Verduyn et al. 1990). The protein concentration in the cell-free extract was determined according to Lowry et al. (1951).
Enzyme assays
These were performed at 30°C with freshly prepared cell-free extracts using a Shimadzu UV-2100 spectrophotometer (Shimadzu, Kyoto, Japan). Reaction rates, corrected for endogenous rates, were proportional to the amounts of extracts added. Glycerol phosphate dehydrogenase (GPDH, EC 1.1.1.8) activity was measured according to Blomberg and Adler (1989). Pyruvate kinase (PYK, EC 2.7.1.40), pyruvate carboxylase (PYC, EC 6.4.1.1), pyruvate decarboxylase (PYDC, EC 6.4.1.1), phosphoenolpyruvate (PEP) carboxykinase (PEPCK, EC 4.1.1.32), and malate synthase (MS, EC 4.1.3.2) activities were measured by the methods of de Jong-Gubbels et al. (1995). PEP carboxylase (PEPC, EC 4.1.1.31) activity was measured by the modified method of Maeba and Sanwall (1969). Phosphoenolpyruvate synthetase (water dikinase pyruvate, PPS, EC 2.7.9.2) activity was measured according to the modified method of Cooper and Kornberg (1969). Alcohol dehydrogenase (ADH, EC 1.1.1.1) and acetaldehyde dehydrogenase (ALD, EC 1.2.1.5 for NAD+, EC 1.2.1.4 for NADP+) activities were measured according to the methods of Postma et al. (1989). Isocitrate dehydrogenase (IDH EC 1.1.1.41) and ribitol dehydrogenase (RDH, EC 1.1.1.56) activities were measured by the methods of Verduyn et al. (1985). AcetylCoA synthase (ACS, EC 6.2.1.1) activity was measured by the method of van den Berg et al. (1996), citrate synthase (CS, EC 4.1.3.7) by the method of de Jong-Gubbels et al. (1996), malate dehydrogenase (MDH, EC 1.1.1.37) by the method of Smith (1983), and malic enzyme (ME, EC 1.1.1.39) by the method of Outlaw and Springer (1983). One unit of activity was defined as the amount of enzyme catalyzing the conversion of 1 μmol substrate per minute. Specific activities were expressed as milliunits per milligram protein. The enzyme activities of a steady-state sample were measured twice using two different aliquots of cell-free extract samples.
Alcohol dehydrogenase and ribitol dehydrogenase activities in the cytosolic membrane of A. aceti were studied by incubating cell membrane fractions discarded from the preparation of cell-free extracts in 5–10 ml of 10 mM maleic acid buffer (pH 7) containing either 70 g/l ethanol or 10 g/l ribitol as a substrate.
Results
Growth experiments
Preliminary growth experiments on Acetobacter aceti IFO 3281 were done in shake flasks on a mineral medium containing ethanol, glycerol, ribitol, xylitol, and their combinations as a carbon source. No growth occurred in a medium containing ribitol or xylitol as the sole source of carbon. Xylitol remained intact in the medium while ribitol was almost fully consumed in 48 h. To our surprise ethanol did not support the growth of A. aceti on a mineral medium but was instead converted into acetic acid.
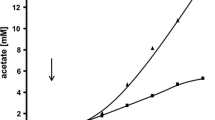
In controlled bioreactor cultivations all the ethanol was converted to acetic acid within 30 h (Fig. 1). Only after 6 days of cultivation did the bacteria start to use the acetic acid in the medium. The utilization of acetate was accompanied by biomass production detected as a change in optical density of the medium, OD600, and as increasing CDW. The final biomass concentration achieved was 0.63 g CDW/l. The biomass yield, YSX, on acetate was 0.04 C-mol/C-mol (0.04 g/g) and the maximum growth rate, μmax, was 0.06 h−1.

Acetobacter aceti IFO 3281 cultivated on a mineral medium containing ethanol as the sole carbon source. Ethanol (black squares), acetic acid (white squares) and biomass (white circles) measured as optical density at 600 nm. The values for OD are multiplied by five
When cultivated on glycerol, the growth started slowly and the exponential phase was reached only after 30 h (Fig. 2). The biomass yield, YSX, was 0.21 C-mol/C-mol (0.16 g/g) and the maximum growth rate, μmax, was 0.15h−1. The final biomass concentration achieved was 1.26 g(CDW)/l. In addition to biomass, 1.1 g/l lactate and 0.1 g/l succinate were produced. The cultivation was finished after the exponential growth phase had ended and the dissolved oxygen value had returned to full saturation (no oxygen consumption). Under these conditions the residual glycerol concentration was 0.45 g/1.

Acetobacter aceti IFO 3281 cultivated on a mineral medium containing glycerol as the sole carbon source. Glycerol (black triangles), lactic acid (white diamonds), succinic acid (crosses) and biomass (white circles) measured as CDW
Growth characteristics in chemostat cultivations
Ethanol, glycerol or an equimolar mixture of ethanol and glycerol were compared as carbon sources in carbon-limited chemostat cultivations on a mineral medium. The dilution rate was 0.036±0.004 h−1, which is well below the μmax for both substrates. The results of the cultivations are presented in Table 1. Biomass and carbon dioxide were the main products of the cultivation, with trace amounts of lactate and succinate accumulating in the medium when glycerol was used as a substrate.
The carbon balance in steady-state samples was 90±2%. Biomass yield was highest on ethanol. The substrate consumption rate was 0.18 C-mmol/C-mmol h−1 for ethanol, 0.24 C-mmol/C-mmol h−1 for glycerol, and 0.22 C-mmol/C-mmol h−1 for the equimolar mixture of ethanol and glycerol. In the mixture the consumption rates of ethanol and glycerol were 0.10 and 0.12 C-mmol/C-mmol h−1, respectively. The oxygen consumption rate per biomass was essentially the same in all the steady-state experiments and thus independent of the substrate used for growth. The carbon dioxide production rate per biomass, on the other hand, was less when A. aceti was cultivated on ethanol than on glycerol or on the mixture of ethanol and glycerol. The respiration quotient was 0.58, 0.78, and 0.80 for ethanol, glycerol, and for the mixture of ethanol and glycerol, respectively. The balances for the degree of reduction were 81–92%.
Enzyme activities
The metabolic pathways used by A. aceti for utilization of ethanol and glycerol were studied by measuring key enzymatic activities in cell-free extracts of the steady-state chemostat samples. The results of the enzymatic assays are presented in Table 2. Very weak soluble alcohol dehydrogenase activity and an NADP/NADPH-dependent acetaldehyde dehydrogenase activity, more than ten times the ADH activity, were detected in all the cell-free extracts.
The AcCoA synthase activity was not found when glycerol was the sole source of carbon and energy. On the other hand, pyruvate decarboxylase was detected only in the cells cultivated on glycerol. Pyruvate carboxylase activity was detected in all the samples.
From the TCA cycle enzymes the activities of isocitrate dehydrogenase and citrate synthase activity were detected in all the samples, but malate dehydrogenase activity was not.
Glyoxylate cycle enzyme, malate synthase, activity was found in the cells grown on ethanol. The activity varied with the amount of ethanol in the medium. In addition, malic enzyme activity was detected in the cells grown on ethanol but not when glycerol was the sole source of carbon. Gluconeogenic enzyme, phosphoenolpyruvate (PEP) carboxylase, adjusting the intracellular levels of oxaloacetic acid and phosphoenolpyruvate, and PEP synthetase converting PEP to pyruvate were present in all the steady-state samples. All the enzyme activities detected, except for PEP synthetase and isocitrate dehydrogenase activity, were of the same order of magnitude.
Bioconversion experiments
Bioconversion experiments in Bioscreen C microcultivation apparatus showed that ribitol was converted to ribulose. Arabitol was converted to xylulose. Conversion rate of xylitol to xylulose was very low. Conversion yield of ribulose was higher in the maleate buffer than in the sodium phosphate buffer.
Bioconversion of ribitol by growing cells of A. aceti in shake flasks was studied in a mineral medium containing ribitol/ethanol or ribitol/glycerol as carbon sources. When ribitol was combined with ethanol, only very little ribulose accumulated in the medium, but all the ethanol was converted to acetic acid. No growth was detected. In a medium containing ribitol and glycerol both of the substrates were simultaneously consumed during the cultivation. Biomass and ribulose were the only products accumulating in the medium.
Ribitol conversion in a chemostat was studied with ribitol as a sole carbon source and as an additional substrate in ethanol- and glycerol-containing media. The dilution rate in the cultivations was 0.045±0.002 h−1. Ribitol was not used for growth and cells were washed out during the chemostat cultivation obeying the theoretical first-order washout kinetics.
When ethanol or glycerol was added as a co-substrate together with ribitol in the medium, only part of the ribitol was converted to l-ribulose while the rest remained intact in the medium. Ethanol and glycerol were fully consumed and no other products than biomass and carbon dioxide were detected. When compared with the cultivations without ribitol, a slight increase in the specific oxygen uptake was noticed (Table 3). The specific carbon dioxide production was approximately the same in the ethanol-containing medium with or without ribitol. The biomass yield was higher when the bacteria were cultivated on a mixture of ethanol and ribitol. In the medium containing ribitol and glycerol, the specific carbon dioxide production was substantially higher and the biomass yield lower compared to the cultivation where no ribitol was present. The respiratory quotient of the cells decreased when ribitol was added to cells growing on ethanol, whereas it increased when ribitol was added into a medium containing glycerol.
The specific conversion activity of ribitol to ribulose was approximately the same regardless of the substrate simultaneously used for growth (Table 3). The specific consumption of ethanol decreased from 4.54 to 4.01 when ribitol was added to the ethanol growing cells, whereas the specific glycerol consumption increased on ribitol addition. As a consequence, the amount of carbon source used per moles of ribitol converted is different for ethanol and glycerol growing cells.
Resting A. aceti cells converted 96% of the 50 g/l ribitol to ribulose with no detectable by-products. The rate of reaction decreased significantly after 24 h when approximately 60% of ribitol was converted (Fig. 3). The activity of the cells was at its maximum 1.0 g g−1 h−1 at 12 h and at its minimum 0.12 g g−1 h−1 at 96 h.

Percentage of ribitol converted to ribulose by resting cells of Acetobacter aceti IFO 3281 in shake flasks containing 50 g/l of ribitol in 50 mM maleate buffer (pH 7)
No oxidation of ethanol or ribitol was detected in cell-free extracts. However, the oxidation activity was in the cell membrane fraction. The rate of ethanol oxidation was 8.7 g l−1 h−1 and that of ribitol 1.7 l−1 h−1.
Discussion
Production of ribulose
l-Ribulose is an important intermediary compound in the pathway to l-ribose – an important starting material for anti-viral drugs. Production of l-ribulose from ribitol is most conveniently done by resting cells of Acetobacter aceti IFO 3281. The reaction first studied by Bhuiyan et al. (1998) was adapted and further studied in our experiments. Contrary to the results of Bhuiyan et al. (1998), the maleate buffer proved to be more suitable for the reaction than the sodium phosphate buffer. In the bioreactor the cells had the initial activity of 1.20 g l-ribulose formed per 1 g of CDW in an hour, which is almost five times higher than the productivity found in test tubes by Bhuiyan et al. (1998).
Ribitol conversion
The ribitol conversion by A. aceti was independent of the growth of the bacterium. Both growing and resting cells were able to convert ribitol to l-ribulose and neither was used in any conditions as a source of carbon or energy. No significant differences in the productivity were detected between growing and resting cells. The dehydrogenase enzyme involved in the conversion is most likely located in the cytoplasmic membrane, since the activity was absent in the cytoplasm, whereas the membrane fraction was able to perform the conversion. Since the reaction has no apparent function in the cellular metabolism, it is assumed that the dehydrogenase enzyme involved is one of the many dehydrogenases found in this strain having an additional activity towards ribitol.
Biomass yield and carbon metabolism
Glycerol was superior as a carbon source for A. aceti. The natural substrate for acetic acid bacteria, namely ethanol, served as a substrate for growth in batch conditions only after extended cultivation times. Only after 6 days incubation was the acetic acid, produced from ethanol, used for growth. In continuous cultivation the situation was different: The highest biomass yield on substrate was achieved when ethanol was the sole carbon source. Ethanol, which is favored as a substrate in the natural habitat of this genus, is not immediately used for growth. In batch cultivation the acetate formed from ethanol is consumed only after the energy reserves of the organism have been exhausted. In chemostat, where the cells are forced to grow, ethanol is used as a growth substrate like glycerol. Ribitol and xylitol were not utilized for growth in any of the studied conditions.
Overall, the biomass yield was low compared with the yields reported for other aerobic bacteria (about 0.5 g/g; Nielsen and Villadsen 1994). A major part of the carbon taken in by the cells of A. aceti was metabolized to CO2. The production of CO2 was accompanied by a significantly high oxygen consumption rate (17.9–18.9 mmol g−1 h−1) compared to the oxygen requirement (8.0 and 10.8 mmol g−1 h−1) reported by Brown (1970) for Saccharomyces cerevisiae and Escherichia coli, respectively.
The fact that biomass yield in chemostat cultivations on glycerol was smaller than that on ethanol might be due to a more energy-requiring pathway. It is feasible that the uptake of glycerol requires energy, whereas ethanol is taken in by passive diffusion. Interestingly, when both ethanol and glycerol were present in the medium, the biomass yield was even more reduced in favor of CO2 production, indicating that the combination of carbon sources was less favorable energetically in providing the precursors for biomass production.
Metabolic pathways
Very few enzyme activities of acetic acid bacteria have been studied and to our knowledge no systematic studies in continuous cultures on metabolic pathways have been published. Most attention has been given to the so-called vinegar-producing enzymes; alcohol dehydrogenase and aldehyde dehydrogenase. Our results of alcohol dehydrogenase and acetaldehyde dehydrogenase activity measurements in the cytoplasm are in accordance with the findings of Nakayama and De Ley (1965). We also tried to detect the activity for glycerol phosphate dehydrogenase in the cell-free extract, but none was found. Since the cells were able to grow on glycerol, they have a pathway for glycerol utilization, and possibly the enzyme is attached to the cytoplasmic membrane.
A. aceti is able to metabolize acetate to carbon dioxide in the reactions of the TCA cycle (Rao 1955; Stanier et al. 1987). Isocitrate dehydrogenase and citrate synthase activities were detected in all the steady-state samples, comparable to the activities reported by Weber (1972), from cultures containing ethanol as the sole carbon source. Malate dehydrogenase activity, on the other hand, was not found in the cytoplasm. According to Benziman and Palgi (1971) the malate dehydrogenase of A. xylinum is NAD-independent, and ferricyanide is the most effective co-factor for both the membrane-bound and soluble malate dehydrogenase found in this strain. This may be the case for malate dehydrogenase in A. aceti as well.
As a result of active respiration, a large amount of carbon dioxide was produced. The production of CO2 per biomass was less with ethanol than with glycerol, probably due to utilization of the glyoxylate cycle where no CO2 is produced. The activity of the glyoxylate cycle enzyme, malate synthase, was dependent on the amount of ethanol in the medium. Additionally malic enzyme activity was detected in ethanol-containing media but not in glycerol media. This is in contradiction to the results of Rao (1955), who did not find malic enzyme activity in A. aceti.
The AcCoA synthase activity, first demonstrated by Rao (1955) and later characterized by O'Sullivan and Ettlinger (1976), was not found when glycerol was the sole carbon source. This explains our observation that no acetate was produced in the glycerol medium in the batch experiments. AcCoA for the TCA cycle was either generated from pyruvate by pyruvate dehydrogenase complex or by some other pathway not yet characterized. On the other hand, pyruvate decarboxylase activity found in the cells grown on glycerol would support acetate formation. In either case, phosphoenolpyruvate must first be converted to pyruvate. This reaction was most likely catalyzed by PEP synthetase since pyruvate kinase was absent, as it also was in the studies of Flückiger and Ettlinger (1977).
In conclusion ethanol was converted to acetate via acetaldehyde followed by conversion of acetate to acetyl coenzyme A. AcCoA then entered the glyoxylate cycle together with oxaloacetate producing energy, reducing power, and precursor metabolites for growth. Oxaloacetate was produced either from malate in the reaction catalyzed by membrane-bound malate dehydrogenase or from pyruvate in the reaction catalyzed by pyruvate carboxylase.
Glycerol was most likely metabolized via glycerol-3-phosphate to dihydroxyacetone phosphate and from there on by the reactions of the EMP pathway to phosphoenolpyruvate. Although conversion of pyruvate to AcCoA has never been demonstrated in acetic acid bacteria, it was assumed that AcCoA for the TCA cycle was either generated from pyruvate by pyruvate dehydrogenase or more likely via acetaldehyde and acetate in the reactions catalyzed by pyruvate decarboxylase, acetaldehyde dehydrogenase, and membrane-bound AcCoA synthetase. Oxaloacetate was produced from pyruvate by pyruvate carboxylase. Schematic presentation of the proposed pathways is shown in Fig. 4.

Schematic presentation of the pathways proposed for utilization of ethanol and glycerol by Acetobacter aceti IFO 3821. Framed boxes show the enzymes studied. The crossed arrow represents an enzyme activity not detected
Since most of the dehydrogenase enzymes catalyzing the oxidation reactions of interest are located in the cytoplasmic membrane, we are presently characterizing these enzymes. Ribitol to ribulose bioconversion rate increased compared with previous studies. More improvements can probably be achieved by careful optimization of biomass concentrations and additives, and by use of biomass recycle and cell revival operations.
References
Adachi O, Fujii Y, Ano Y, Moonmangmee D, Toyama H, Shinagawa E, Theergool G, Lotong N, Matsuhita K (2001) Membrane-bound sugar alcohol dehydrogenase in acetic acid bacteria catalyzes l-ribulose formation and NAD-dependent ribitol dehydrogenase is independent of the oxidative fermentation. Biosci Biotech Biochem 65:115–125
Benziman M, Palgi A (1971) Characterisation and properties of the pyruvate phosphorylation system of Acetobacter xylinum. J Bacteriol 108:211–218
Berg MA van den, Jong-Gubbels P de, Kortland CJ, Dijken JP van, Pronk JT, Steensma HY (1996) The two acetyl-coenzyme A synthetases of Saccharomyces cerevisiae differ with respect to kinetic properties and transcriptional regulation. J Biol Chem 271:28953–28959
Bhuiyan SH, Ahmed Z, Utamura M, Izumori K (1998) A new method for the production of l-lyxose from ribitol using microbial and enzymatic reactions. J Ferment Bioeng 86:513–516
Blomberg A, Adler L (1989) Roles of glycerol and glycerol-3-phosphate dehydrogenase (NAD+) in acquired osmotolerance of Saccharomyces cerevisiae. J Bacteriol 171:1087–1092
Brown DE (1970) Aeration in the submerged culture of microorganisms. (Methods in microbiology vol 2) Academic Press, New York, pp 125–174
Cooper RA, Kornberg HL (1969) Phosphoenolpyruvate synthetase. (Methods in enzymology vol XIII) Academic Press, New York, pp 309–314
Flückiger R, Ettlinger L (1977) Glucose metabolism in Acetobacter aceti. Arch Microbiol 114:183–187
Jokela J, Pastinen O, Leisola M (2001) Isomerization of pentose and hexose sugars by an enzyme reactor packed with cross-linked xylose isomerase crystals. Enzyme Microb Technol 31:67–76
Jong-Gubbels P de, Vanrolleghem P, Heijnen S, Dijken JP van, Pronk JT (1995) Regulation of carbon metabolism in chemostat cultures of Saccharomyces cerevisiae grown on mixtures of glucose and ethanol. Yeast 11:407–418
Jong-Gubbels P de, Dijken JP van, Pronk JT (1996) Metabolic fluxes in chemostat cultures of Schizosaccharomyces pombe grown on mixtures of glucose and ethanol. Microbiol 142:1399–1407
Leisola M, Jokela J, Finell J, Pastinen O (2001) Simultaneous catalysis and product separation by cross-linked enzyme crystals. Biotechnol Bioeng 72:501–505
Lowry OH, Roseborough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265 −275
Maeba P, Sanwall BD (1969) Phosphoenolpyruvate carboxylase from Salmonella typhimurium, strain LT2. (Methods in enzymology vol XIII) Academic Press, New York, pp 283–288
Matsuhita K, Takaki Y, Shinagawa E, Ameyama M, Adachi O (1992) Ethanol oxidase respiratory chain of acetic acid bacteria: reactivity with ubiquinone of pyrroloquinoline quinone-dependent alcohol dehydrogenase purified from Acetobacter aceti and Gluconobacter suboxydans. Biosci Biotechnol Biochem 56:304–310
Nakayama T (1961a) Studies on acetic acid bacteria. III. Purification and properties of coenzyme-independent aldehyde dehydrogenase. J Biochem 49:158–163
Nakayama T (1961b) Studies on acetic acid bacteria. IV. Purification and properties of a new type of alcohol dehydrogenase, alcohol sytochrome-553 reductase. J Biochem 49:240–251
Nakayama T, De Ley J (1965) Localisation and distribution of alcohol-cytochrome 553 reductase in acetic acid bacteria. Antonie van Leeuwenhoek 31:205–219
Neidhardt FC, Ingraham JL, Schaechter M (1990) Physiology of the bacterial cell. Sinauer Associates, Sunderland, Mass.
Nielsen J, Villadsen J (1994) Bioreactor engineering principles. Plenum, New York, p 173
O'Sullivan J, Ettlinger L (1976) Characterization of the acetyl-CoA synthetase of Acetobacter aceti. Biochim Biophys Acta 450:410–417
Outlaw WH Jr, Springer SA (1983) 'Malic' enzyme. (Methods of enzymatic analysis vol III) VCH, Weinheim, pp 176–182
Pastinen O, Visuri K, Schoemaker H, Leisola M (1999) Novel reactions of xylose isomerase from Streptomyces rubiginosus. Enzyme Microb Technol 25:695–700
Postma E, Verduyn C, Scheffers WA, Dijken JP van (1989) Enzymic analysis of the Crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microbiol 55:468–477
Rao RMR (1955) Pyruvate and acetate metabolism in Acetobacter suboxydans and Acetobacter aceti. Dissertation, University of Illinois, Urbana, Ill.
Reichstein T, Grussner A (1934) Eine ergiebige synthese der l-ascorbinsaure (C-vitamin). Helv Chim Acta 17:996
Scott A (2002) Danisco aims to ramp up sales of 'Pharmaceutical' sugars. Chem Week Nov 20–Nov 27
Smith AF (1983) Malate dehydrogenase. (Methods of enzymatic analysis vol III) VCH, Weinheim, pp 163–176
Stanier RY, Ingraham JL, Wheelis ML, Painter PR (1987) The acetic acid bacteria. In: General microbiology, 5th edn. Macmillan Education, Hong Kong, pp 417–419
Verduyn C (1991) Energetic aspects of metabolic fluxes in yeasts. Dissertation, Delft University of Technology, Delft, The Netherlands
Verduyn C, Kleef R van, Frank J, Schreuder H, Dijken JP van, Scheffers WA (1985) Properties of the NAD(P)H-dependent xylose reductase from the xylose-fermenting yeast Pichia stipitis. Biochem J 226:669–677
Verduyn C, Postma E, Scheffers WA, Dijken JP van (1990) Physiology of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. J Gen Microbiol 136:395–403
Weber P (1972) Der Einfluss von Glucose auf den Aethanolstoffwechsel eines Stammes von Acetobacter aceti. Dissertation, ETH-Zurich, Switzerland
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kylmä, A.K., Granström, T. & Leisola, M. Growth characteristics and oxidative capacity of Acetobacter aceti IFO 3281: implications for l-ribulose production. Appl Microbiol Biotechnol 63, 584–591 (2004). https://doi.org/10.1007/s00253-003-1406-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1406-4