
Abstract
A novel enzyme, d-3-hydroxyaspartate aldolase (d-HAA), catalyzing the conversion of d-3-hydroxyaspartate to glyoxylate plus glycine, was purified to homogeneity from Paracoccus denitrificans IFO 13301. d-HAA is strictly d-specific as to the α-position, whereas the enzyme does not distinguish between threo and erythro forms at the β-position. In addition to d-3-hydroxyaspartate, the enzyme also acts on d-threonine, d-3-3,4-dihydroxyphenylserine, d-3-3,4-methylenedioxyphenylserine, and d-3-phenylserine. The d-HAA gene was cloned and sequenced. The gene contains an open reading frame consisting of 1,161 nucleotides corresponding to 387 amino acid residues. The predicted amino acid sequence displayed 35% and 22% identity with that of the d-threonine aldolase of Arthrobacer sp. DK-38 and Alcaligenes xylosoxidan IFO 12669, respectively. This is the first paper reporting both a purified enzyme with d-3-hydroxyaspartate aldolase activity and also its gene cloning.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
3-Hydroxyaspartate and its derivatives have attractive functions, acting as antimicrobial (Ishiyama et al. 1975) and competitive blockers of the excitatory glutamate/aspartate transporters of the mammalian nervous system (Balcar et al. 1977; Lebrun et al. 1997). However, the chemical synthesis of 3-hydroxyaspartate requires multiple steps to isolate its four isomers (the l-threo, d-threo, l-erythro, and d-erythro forms) (Kaneko and Katsura 1963; Shimamoto 2000). We have studied various threonine aldolases with different substrate specificities (Liu et al. 2000a), but none showed activity toward 3-hydroxyaspartate. 3-Hydroxyaspartate aldolase (EC 4.1.3.14), which stereospecifically catalyzes the retro-aldol cleavage of erythro-3-hydroxyaspartate, is a potentially useful catalyst for the optical resolution of 3-hydroxyaspartate and its derivates. Gibbs and Morris (1964) reported 3-hydroxyaspartate aldolase activity in the cell extract of Paracoccus denitrificans NCIMB 8944. However, the enzyme was neither purified nor well characterized.
This paper describes the purification and characterization of d-3-hydroxyaspartate aldolase from Paracoccus denitrificans IFO 13301, and its gene cloning, sequencing, and expression in E. coli cells. This is the first report of a purified enzyme with d-3-hydroxyaspartate aldolase activity, providing a new process for producing an optically pure hydroxyaspartate.
Materials and methods
Materials
DEAE-Toyopearl and Phenyl-Toyopearl were purchased from Tosoh (Tokyo, Japan); Gigapite (hydroxyapatite) was bought from Toagosei Chemical Industry (Tokyo, Japan); HiLoad Superdex 200 and Mono Q were from Pharmacia Biotech (Uppsala, Sweden). dl-threo-3-hydroxyaspartate was synthesized by the ring-opening reaction of cis-epoxysuccinic acid, as has been described previously (Kaneko and Katsura 1963); dl-erythro-, d- and l-threo isomers of 3-hydroxyaspartate were purchased from Wako Pure Chemicals (Osaka, Japan). dl-3-3,4-Methylenedioxyphenylserine was prepared according to the method of Ohashi et al. (1984). The other chemicals were all analytical-grade reagents.
Bacterial strains, plasmids, and culture conditions
Paracoccus denitrificans IFO 13301 was used as the source of chromosomal DNA (Saito and Miura 1963). Escherichia coli XL1-Blue MRF′ [recA1 thi endA1 supE44 gyrA46 relA1 hsdR17 lac/F′ (proAB + lacI q lacZ∆ M15::Tn10 Tetr)] (Toyobo, Osaka, Japan) was used as a host for the gene cloning. Cosmid pWE15 (Stratagene, La Jolla, Calif.) was used as a vector for the construction of the gene library. Plasmid pUC118 (Takara Shuzo, Kyoto, Japan) was used as a vector for the cloning of the PCR product. pKK 223-3 (Pharmacia Biotech) was used as a vector for overexpression of the d-HAA gene. Paracoccus denitrificans IFO 13301 was grown at 30°C in a medium comprising 0.5% peptone, 0.05% yeast extract, and 0.5% glycolate and 0.1% magnesium sulfate, pH 7.0. Recombinant E. coli cells were cultivated at 37°C in Luria-Bertani (LB) medium (1% peptone, 0.5% yeast extract, and 1% NaCl, pH 7.2) containing 0.1 mg/ml of ampicillin. To induce the expression of the gene under the control of the tac promoter, 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) was added to the LB medium.
General recombinant DNA technique
Plasmid DNA was purified with a plasmid purification kit from Qiagen (Chatsworth, Calif.). Restriction enzymes and DNA-modifying enzymes were purchased from Takara Shuzo and Toyobo and used according to the manufacturers' directions. Transformation of E. coli with plasmid DNA by electroporation was performed under standard conditions with a BTX ECM 600 electroporation system (Biotechnologies and Experimental Research, San Diego, Calif.). Other general procedures were performed as described by Sambrook et al. (1989).
d-HAA purification
All enzyme purification procedures were carried out at 0–5°C. 50 mM potassium phosphate, pH 7.0, containing 10 μM PLP was used as the buffer throughout the purification unless otherwise stated.
Step 1: preparation of cell-free extract
The cells of Paracoccus denitrificans IFO 13301, grown aerobically at 30°C for 18 h in 50 l of the medium described above, were harvested and rinsed with the buffer. After being suspended in 500 ml of the buffer, the cells were disrupted by ultrasonic oscillation at 4°C for 20 min with a model 201 M ultrasonic oscillator (Kubota, Tokyo, Japan). The cell debris was removed by centrifugation at 25,000 g for 30 min.
Step 2: ammonium sulfate fractionation
The supernatant solution was fractionated with ammonium sulfate from 30% to 70% saturation. The precipitate collected by centrifugation at 25,000 g for 30 min was dissolved in the buffer.
Step 3: DEAE-Toyopearl column chromatography
The enzyme solution was dialyzed against 1,000 vol. of the buffer and applied to a DEAE-Toyopearl 650 M column (5×40 cm) equilibrated with the buffer. After the column had been washed thoroughly with the buffer containing 50 mM NaCl, a 3,000-ml linear gradient elution was performed with the buffer supplemented with NaCl by increasing the concentrations from 50 mM to 300 mM. The flow rate was maintained at 5 ml/min. The active fractions were pooled and concentrated by ultrafiltration with a Centriprep-30.
Step 4: Gigapite (hydroxyapatite) column chromatography
The enzyme solution was dialyzed against 1,000 vol. of the buffer and applied to a Gigapite column (5×40 cm) equilibrated with the buffer. After the column had been washed thoroughly with the buffer, a 3,000-ml linear gradient elution was performed with the buffer by increasing the concentration from 50 mM to 300 mM. The flow rate was maintained at 5 ml/min. The active fractions were pooled and concentrated by ultrafiltration.
Step 5: HiLoad Superdex gel filtration
The enzyme solution was applied to a HiLoad Superdex 200 column (1.6×60 cm) equipped with a Pharmacia FPLC system, and then eluted with the buffer at a flow rate of 1 ml/min. The active fractions were pooled and concentrated by ultrafiltration.
Step 6: Phenyl-Toyopearl column chromatography
The supernatant solution, brought to 30% saturation with ammonium sulfate, was applied to a Phenyl-Toyopearl column (2.5×20 cm). After the column had been washed thoroughly with the buffer containing 30% ammonium sulfate, elution was carried out with a 500-ml linear gradient of 30–0% saturated ammonium sulfate in the buffer at a flow rate of 5 ml/min, and the active fractions were pooled and concentrated by ultrafiltration.
Step 7: Mono Q chromatography
The enzyme solution was dialyzed against 1,000 vol. of the buffer and applied to a Mono Q HR 10/10 column (1×5 cm) equipped with a FPLC system. After the column had been washed thoroughly with the buffer, elution was carried out with a 50 ml linear gradient of 50–300 mM NaCl in the buffer at a flow rate of 1.0 ml/min. The fractions, showing single bands on SDS-PAGE and aldolase activity, were pooled and concentrated by ultrafiltration with a Centriprep-30 (Amicon, Beverly, Mass.). The purified enzyme was stored in buffer containing 20% (w/v) glycerol at −30°C.
Cloning of the d-HAA gene (dhaa)
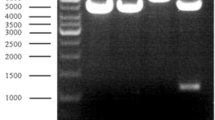
Two oligonucleotide primers were made-to-order by Hokkaido System Science (Sapporo, Japan), each with additional restriction sites (underlined in the following sequences) added to the 5′ end to facilitate cloning of the amplified product: primer I, 5′-CCGAAGCTTATGAAYGCNAARAC-3′; and primer II, 5′-GCCGAATTCTCCATRCCNGGYAG-3′. Degenerate positions are indicated by "Y" for C or T, "R" for A or G, and "N" for all bases. Primers I and II were based upon the NH2-terminal amino acid sequence (Met-Asn-Ala-Lys-Thr-Asp-Phe-Ser-Gly-Tyr-Glu-Val-Gly-Tyr-Asp-Ile-Pro-Ala-Leu-Pro-Gly-Met-Asp) of the wild-type d-HAA from Paracoccus denitrificans IFO 13301 (see below). Polymerase chain reaction amplification was performed in 100 µl of 10 mM Tris-HCl (pH 8.85), 25 mM KCl, 5 mM (NH4)2SO4, 2 mM MgSO4, 0.1 mM dNTP, 20 pmol of each primer, 1 µg of the genomic DNA and 1 U of PWO DNA polymerase (Boehringer, Mannheim, Germany) at 94°C for 1 min, 43°C for 0.5 min, and 72°C for 1 min in a total of 30 cycles. The amplified product was digested with EcoRI and HindIII, separated by agarose gel electrophoresis, and then purified with a GeneClean kit (Bio101, Vista, Calif.). The amplified DNA of about 80 bp was then cloned into pUC118. Chromosomal DNA isolated from Paracoccus denitrificans IFO 13301 cells was partially digested with Sau3AI and fractionated on a sucrose density gradient (10–40%) in a Beckman L-70 ultracentrifuge at 100,000 g for 18 h. The fragments in the molecular weight range of 30–50 kb were collected and ligated into BamHI-restricted pWE15. This concatemer was packaged in vitro into bacteriophage λ particles using an in vitro packaging kit (Gigapack III Gold Packaging Extract; Stratagene) and used to infect E. coli XL1-Blue MRF′. The genomic library was screened by colony hybridization with the (α-32P)dATP-labeled 80-bp DNA fragment as a probe. The clone, pWDHAA, carrying an approximately 35-kb DNA fragment, was selected for further analysis.
Nucleotide sequence analysis
pWDHAA was used as a sequencing template. The nucleotide sequence was determined by the dideoxy chain termination method with Cy5 AutoCycle sequencing kits, and a Pharmacia LKB ALFred DNA sequencer. A homology search was performed by means of the sequence similarity searching programs, Fasta (Pearson and Lipman 1988), and Blast (Altschul et al. 1990). The Clustal W method was used to align the sequences (Higgins et al. 1996).
Expression of the dhaa gene in E. coli cells
To obtain the entire gene without excessive flanking parts, PCR amplification was carried out in a 50-µl reaction mixture containing 5 μl of DMSO, 10 mM Tris-HCl (pH 8.85), 25 mM KCl, 5 mM (NH4)2SO4, 2 mM MgSO4, 0.1 mM deoxynucleotide triphosphate, 20 pmol of each primer, 1 µg of the genomic DNA, and 0.5 U of PWO DNA polymerase (Boehringer) at 94°C for 1 min, 60°C for 2 min, and 72°C for 3 min for a total of 30 cycles. The 5′ primer containing a Shine-Dalgarno sequence (lower-case letters) and an ATG initiation codon (bold letters), and the 3′ primer with the complement of the TGA termination codon (bold letters) had the respective sequences 5′-GCCGAATTCggagAGCCCCATGAATGC-3′ and 5′-CCGGAATTCCGACGAGCATCAGTAGC-3′, which were designed on the basis of the nucleotide sequence of the dhaa gene; to facilitate the cloning, an additional restriction site (underlined sequence) was incorporated into both primers. The amplified PCR product was digested with EcoRI, separated by agarose gel electrophoresis, and then purified with a GeneClean kit. The amplified DNA of approximately 1.1 kb, which contained the complete coding sequences of the dhaa gene, was inserted downstream of the tac promoter in pKK223-3 and then used to transform E. coli XL1-Blue MRF′ cells. The constructed plasmid was designated pDHAA.
Molecular mass determination and amino acid sequencing
The molecular mass of the enzyme was determined by gel filtration on a TSK gel G3000 SWXL column (7.5×600 mm) (Tosoh) with the following marker proteins: glutamate dehydrogenase (290 kDa), lactate dehydrogenase (140 kDa), enolase (67 kDa), adenylate kinase (32 kDa), and cytochrome c (12.4 kDa) (Wako Pure Chemicals). The subunit molecular mass of the enzyme was determined by SDS-PAGE with the following marker proteins: phosphorylase b (94 kDa), albumin (67 kDa), ovalbumin (43 kDa), carbonic anhydrase (30 kDa), trypsin inhibitor (20.1 kDa), and α-lactalbumin (14.4 kDa) (Pharmacia). The NH2-terminal amino acid sequence was determined by the Edman degradation procedure with a 476A protein sequencer (Perkin-Elmer, Norwalk, Conn.).
Enzyme and protein assays
Hydroxyaspartate aldolase activity was assayed with dl- erythro-3-hydroxyaspartate as a substrate. The reaction mixture comprised 10 μmol of dl- erythro-3-hydroxyaspartate, 0.01 μmol of pyridoxal phosphate (PLP), 1 μmol of magnesium sulfate, 20 μmol of N-(2-hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer, pH 8.0, and the enzyme, in a total volume of 200 μl. The reaction was carried out at 30°C for 10 min and was terminated by the addition of 66 μl of 2,4-dinitrophenylhydrazine reagent. After a further incubation at room temperature for 20 min, 334 μl of 10% NaOH was added, and the developed color was measured spectrophotometrically at 454 nm. One unit of the enzyme was defined as the amount which catalyzed the formation of 1 μmol of glyoxylate per minute under the assay conditions described. The threonine aldolase, phenylserine aldolase, and methylenedioxyphenylserine aldolase activities were measured spectrophotometrically as described previously (Liu et al. 1998, 2000b).
Protein concentration was determined with a Bio-Rad protein assay kit using bovine serum albumin as the standard.
Chromatographic optical resolution of amino acid enantiomers
The isomers of hydroxyaspartate was derived with o-phthaldialdehyde and analyzed by HPLC on a column of 5C18-MS (0.46×25 cm) (Nacalai, Kyoto, Japan) with 48 mM sodium phosphate buffer (pH 6.5) containing 2% methanol and 2% tetrahydrofuran as the solvent; the flow rate was 0.6 ml/min, detection was at 340 nm and temperature was 40°C. The isomers of phenylserine and methylenedioxyphenylserine were also analyzed by HPLC as follows: column, Sumichiral OA-5000 (0.46×15 cm) (Sumitomo, Tokyo, Japan); solvent, 2 mM copper sulfate containing 15% methanol; flow rate, 1.0 ml/min; detection, 254 nm; and temperature, 30°C.
Nucleotide sequence accession number
The nucleotide sequence reported in this paper will appear in the DDBJ, EMBL, and GenBank nucleotide sequence databases under accession number AB075600.
Results
d-HAA purification
d-HAA from Paracoccus denitrificans IFO 13301 was purified by ammonium sulfate fractionation, DEAE-Toyopearl, Gigapite, HiLoad Superdex gel filtration, Phenyl-Toyopearl and Mono Q chromatographies (see Table 1). About 5.4 mg of purified enzyme was obtained from 150 g of wet cells.
Enzyme characterization
Molecular mass
The purified enzyme showed a single protein band on SDS-PAGE with a molecular mass of about 43 kDa (Fig. 1). The native molecular mass of purified d-HAA was determined to be 80 kDa by gel filtration. These results suggest that d-HAA from Paracoccus denitrificans IFO 13301 exists as a dimer.

Purification of d-3-hydroxyaspartate aldolase from Paracoccus denitrificans IFO 13301. The purified enzyme was loaded on a 10% SDS-polyacrylamide gel and stained with Coomassie Blue after electrophoresis. Lane 1, the purified enzyme (10 μg); Lane 2, molecular mass standards. The numbers to the left are the molecular masses of the standards
Cofactor requirement
The enzyme following sufficient dialysis without PLP exhibited absorption maxima at 280 and 419 nm, with an A280/A419 ratio of about 5.8 (data not shown). Solutions of the pure enzyme are distinctly yellow in color. These results suggested that the enzyme contains PLP as a prosthetic group. To confirm this, the holoenzyme was converted to the apoenzyme by treatment with 1 mM hydroxylamine at 25°C for 30 min and then dialyzed against 20 mM potassium phosphate buffer (pH 7.0). The constructed apoenzyme did not show d-3-hydroxyaspartate aldolase activity, but the activity was restored to 83% of that of the native enzyme on incubation with 0.1 mM PLP.
In addition, the enzymic activity was found to be strongly inhibited by 1 mM EDTA (82% inhibition), suggesting that metal ions were involved in the reaction. To study the effect of metal ions on the enzymic activity, the enzyme was treated with 1 mM EDTA at 25°C for 60 min and then dialyzed against 50 mM TRIS-HCl buffer (pH 7.4) containing 10 μM PLP. The specific activity of the EDTA-treated enzyme was 5.4 U/mg. When 1 mM MnCl2, MgCl2, and CoCl2 were added, the activity increased to 43.7, 29, and 10.8 U/mg, respectively. In contrast, KCl and NaCl did not have an effect on the enzymic activity.
pH and temperature effects
To examine the effect of pH on the enzymic activity, the initial reaction velocity was measured by the standard assay method using dl- erythro-3-hydroxyaspartate as substrate with the following buffers of various pH values: 2-(N-morpholino)ethanesulfonic acid (pH 5.5–6.5), HEPES (pH 7.0–8.0), and 1,3-bis[tris(hydroxymethyl)methylamino]propane (pH 7.0–11). The maximum activity of the d-HAA was found to be at pH 9.0 (Fig. 2). The enzyme was stable between pH 6.5 and 8.5 for 30 min at 30°C. The effect of temperature was also examined. The maximum activity of d-HAA was observed at 35°C, and the enzyme retained 50% activity upon heating at 45°C for 30 min.

Effect of pH on d-3-hydroxyaspartate aldolase from Paracoccus denitrificans IFO 13301. d-3-Hydroxyaspartate aldolase activity was determined with dl- erythro-3-hydroxyaspartate as a substrate, according to the method described in the Materials and methods section
Substrate specificity
The substrate specificity of the enzyme is shown in Table 2. The enzyme acted on both d-erythro-3-hydroxyaspartate and d-threo-3-hydroxyaspartate, but was inert toward their l- counterparts. The V max and K m of the enzyme toward d-erythro-3-hydroxyaspartate were determined to be 30 U/mg and 0.4 mM, respectively, under standard assay conditions. The enzyme showed a broad substrate specificity. In addition to 3-hydroxyaspartate, d-allo-threonine, d-threonine, d-threo-3-phenylserine, d-erythro-3-phenylserine, d-erythro-3-3,4-methylenedioxyphenylserine, and d-threo-3-3,4-methylenedioxyphenylserine were also found to be substrates of the enzyme (Table 2).
Amino acid sequencing
The NH2-terminal amino acid sequence of the enzyme was determined to be Met-Asn-Ala-Lys-Thr-Asp-Phe-Ser-Gly-Tyr-Glu-Val-Gly-Tyr-Asp-Ile-Pro-Ala-Leu-Pro-Gly-Met-Asp.
Cloning of the dhaa gene
The primers used for cloning of the dhaa gene by PCR were based on the NH2-terminal amino acid sequence of the purified d-HAA from Paracoccus denitrificans IFO 13301. PCR with the primers and the bacterial chromosomal DNA as the template yielded an amplified band of about 80 bp. The amplified DNA was then cloned into pUC118 in E. coli. Nucleotide sequencing of the 80 bp fragment showed the presence of an open reading frame (ORF) continuing over the entire sequence. The deduced amino acid sequence of the PCR fragment was in perfect agreement with the NH2-terminal amino acid sequence determined from the purified d-HAA from Paracoccus denitrificans IFO 13301. We then directly performed colony hybridization with the 80-bp fragment as a probe using the established genomic library of Paracoccus denitrificans IFO 13301. Two positive recombinant E. coli clones were obtained from about 1,000 transfectants. One of the clones harboring a smaller DNA fragment insert (about 35-kb) was chosen for further characterization.
Sequence analysis of the dhaa gene
The plasmid, pWDHAA, extracted from the positive clone, was directly used as the template for sequencing the dhaa gene by the gene-walking method; the initial sequencing primer was designed based on the nucleotide sequence of the 80-bp PCR product.
Sequence analysis of the double-strand DNA revealed that the ORF consists of 1,164 bp starting with an initiation codon, ATG, and ending with a termination codon, TGA (see Fig. 3). A probable ribosome-binding sequence, GGAG, is present six bases upstream of the putative translational start codon. The ORF encodes a protein of 387 amino acid residues. The predicted molecular weight is 41,638, which is in good agreement with the value determined for the purified enzyme from Paracoccus denitrificans IFO 13301. The NH2-terminal amino acid sequence coincided with that of the purified enzyme determined by Edman degradation (Fig. 3).

Nucleotide and deduced amino acid sequences of the dhaa gene and flanking regions. A putative Shine-Dalgarno sequence is double underlined. The asterisk denotes a translational stop codon. The NH2-terminal amino acid sequence coinciding with that of the purified enzyme determined by Edman degradation is underlined
Expression of the dhaa gene in E. coli cells
The whole dhaa gene amplified by PCR directly from the bacterial chromosomal DNA, with a putative Shine-Dalgarno sequence (GGAG) and an initiation codon (ATG), was inserted into the EcoRI site of pKK223-3. The resultant plasmid pKDHAA was introduced into E. coli XL1-Blue MRF′ cells. The nucleotide sequence of the whole amplified gene was further confirmed to have undergone no error matching during the PCR by sequencing of the double strands. The recombinant cells produced a large amount of d-HAA, and the specific activity of the crude extract of E. coli XL1-Blue harboring pKDHAA was elevated to 5.8 U/ mg. The protein was only produced in the presence of IPTG (data not shown), indicating that the tac promoter is essential for the overexpression.
Sequence homology with other proteins
In a search of protein amino acid databases, the predicted amino acid sequence was shown to have 35% and 22% identity with that of the d-threonine aldolase of Arthrobacer sp. DK-38 and Alcaligenes xylosoxidan IFO 12669, respectively. In addition, the enzyme was found to have 16, 15, 18, 25, 30, 33, and 34% identity in primary structure to d-serine deaminase (GenBank, U41162) of Burkholderia cepacia and six hypothetical proteins of Shewanella sp. strain SCRC-2738 (GenBank, U73935), Corynebacterium glutamicum (GenBank, AX063849), Mesorhizobium loti (GenBank, AP003013), Agrobacterium tumefaciens (GenBank, AE009323), Sinorhizobium meliloti (Genbank, AL603644), and Caulobacter crescentus (Genbank, AE005974), respectively.
Discussion
Gibbs and Morris (1964) reported 3-hydroxyaspartate aldolase activity in cell extract of Paracoccus denitrificans NCIMB 8944. However, the enzyme had been neither purified nor well characterized. To understand the biochemistry of the enzyme and obtain a biocatalyst for production of an optical pure 3-hydroxyaspartate, we have succeeded in the purification of a d-HAA from Paracoccus denitrificans IFO 13301. The purified enzyme, requiring PLP and divalent cations as cofactors for maximal activity, was found to stereospecifically catalyze the cleavage of both d-erythro- and d-threo-3-hydroxyaspartate to glycine and glyoxylic acid. This is the first purified enzyme showing activity to cleave d-β-hydroxyaspartate.
3-Hydroxyaspartate pathway, shown in Fig. 4, was reported to play an essential role in the growth of Paracoccus denitrificans on glycolate or other precursors of glyoxylate (Gibbs and Morris 1965; Kornberg and Morris 1965). 3-Hydroxyaspartate aldolase, catalyzing the formation of hydroxyaspartate from glycine and glyoxylate, was proposed to be one of the key enzymes for this pathway (Gibbs and Morris 1964; Kornberg and Morris 1965). However, the purified d-HAA failed to catalyze the aldol reaction from glycine and glyoxylate under any of the conditions investigated (data not shown). We assume that either the isolated protein is different from the one reported by Gibbs and Morris previously, or we have not found favorable in vitro aldol reaction conditions for the enzyme in a pure form. We are attempting to clone the other two genes encoding glyoxylate-l-aspartate aminotransferase and 3-hydroxyaspartate dehydratase of the proposed 3-hydroxyaspartate pathway for more detailed genetic studies of this unique microbial metabolism.

Proposed 3-hydroxyaspartate pathway of Paracoccus denitrifican (Kornberg and Morris 1965). 1. Glyoxylate-l-aspartate aminotransferase; 2. 3-hydroxyaspartate aldolase; 3. 3-hydroxyaspartate dehydratase
In previous work, we identified that Lys59 of d-threonine aldolase from Arthrobacter sp. DK-38, corresponding to Lys62 of d-HAA, was the pyridoxal binding site of the enzyme by chemical modification with NaBH4 (Liu et al. 1998). In this study, we found that d-HAA had rather high amino acid sequence similarity with d-threonine aldolases of Arthrobacer sp. DK-38 (Liu et al. 1998) and Alcaligenes xylosoxidan IFO 12669 (Liu et al. 2000b), d-serine deaminase of Burkholderia cepacia, and six hypothetical proteins (see above). Sequence alignment showed that Lys62 of d-HAA was completely conserved among all these proteins (data not shown). It is very likely that Lys62 of d-HAA plays a role as the pyridoxal binding site of the enzyme for the retro-aldol reaction.
From an enzymological point of view, it would be interesting to compare the three-dimensional structures of d-HAA and d-threonine aldolases, which showed high amino acid sequence similarity but different substrate specificities, to elucidate the structural and functional relationship of the enzymes.
From the point of view of applications, like threonine aldolases (Liu et al. 2000a), d-HAA would be a useful catalyst for resolving racemic 3-hydroxyaspartate to give l-3-hydroxyaspartate and other β-hydroxy-α-amino acids.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Balcar VJ, Jonston GA, Twitchin B (1977) Stereospecificity of the inhibition of l-glutamate and l-aspartate high affinity uptake in rat brain slices by threo-3-hydroxyaspartate. J Neurochem 28:1145–1146
Gibbs RG, Morris JG (1964) Assay and properties of erythro-β-hydroxyasparte aldolase from Micrococcus denitrificans. Biochim Biochem Acta 85:501–503
Gibbs RG, Morris JG (1965) Purification and properties of erythro-β-hydroxyasparte dehydratase from Micrococcus denitrificans. Biochem J 97:547–554
Higgins DG, Thompson JD, Gibson TJ (1996) Using CLUSTAL for multiple sequence alignments. Methods Enzymol 66:383–402
Ishiyama T, Furuta T, Takai M, Okimoto Y (1975) l- threo-β-hydroxyaspartate acid as an antibiotic amino acid. J Antibiotics 23:821–823
Kaneko T, Katsura H (1963) The synthesis of four optical isomers of β-hydroxyaspartatic acid. Bull Chem Soc Jpn 36:899–930
Kornberg HL, Morris JG (1965) The utilization of glycollate by Micrococcus denitrificans: the β-hydroxyaspartate pathway. Biochem J 95:577–586
Lebrun B, Sakitani M, Shimamoto K, Yasuda-Kamatani Y, Nakajima T (1997) New β-hydroxyaspartate derivatives are competitive blockers for the bovine glutamate/aspartate transporter. J Biol Chem 272:20336–20339
Liu JQ, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H (1998) Novel metal-activated pyridoxal enzyme with a unique primary structure, low-specificity d-threonine aldolase from Arthrobacter sp. strain DK-38. J Biol Chem 273:16678–16685
Liu JQ, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H (2000a) Diversity of microbial threonine aldolasees and their application. J Mol Cat B Enzymatic 10:107–115
Liu JQ, Otani M, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H (2000b) Gene cloning and overproduction of low-specificity d-threonine aldolase from Alcaligenes xylosoxidans and its application for production of a key intermediate for parkinsonism drug. Appl Microbiol Biotechnol 54:44–51
Ohashi N, Nagata S, Ishizumi K, Maeshima K (1984) European Patent 83,300,059
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85:2444–2448
Saito H, Miura K (1963) Preparation of transforming deoxyribonucleic acid by phenol treatment. Biochim Biophys Acta 72:619–629
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, Lebrun B, Yumoto N, Nakajima T (2000) Syntheses of optically pure β-hydroxyaspartate derivatives as glutamate transporter blockers. Bioorg Med Chem Lett 10:2407–2410
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, J.Q., Dairi, T., Itoh, N. et al. A novel enzyme, d-3-hydroxyaspartate aldolase from Paracoccus denitrificans IFO 13301: purification, characterization, and gene cloning. Appl Microbiol Biotechnol 62, 53–60 (2003). https://doi.org/10.1007/s00253-003-1238-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-003-1238-2