
Abstract
Knowledge about the magnitude of individual polymorphism is a critical part in understanding the complexity of comprehensive mismatching. HLA-B*44:09 differs from the highly frequent HLA-B*44:02 allele by amino acid exchanges at residues 77, 80, 81, 82 and 83. We aimed to identify the magnitude of these mismatches on the features of HLA-B*44:09 bound peptides since residues 77, 80 and 81 comprise part of the F pocket which determines sequence specificity at the pΩ position of the peptide. Using soluble HLA technology we determined >200 individual (nonduplicate) self-peptides from HLA-B*44:09 and compared their features with that of the published peptide features of HLA-B*44:02. Both alleles illustrate an anchor motif of E at p2. In contrast to the C-terminal peptide binding motif of B*44:02 (W, F, Y or L), B*44:09-derived peptides are restricted predominantly to L or F. The source of peptides for both alleles is identical (LCL 721.221 cells) allowing us to identify 23 shared peptides. The majority of these peptides however contained the restricted B*44:09 anchor motif of F or L at the pΩ position. Molecular modelling based on the B*44:02 structure highlights that the differences of the C-terminal peptide anchor between both alleles can be explained primarily by the B*44:0281Ala > B*44:0981Leu polymorphism which restricts the size of the amino acid that can be accommodated in the F pocket of B*44:09. These results highlight that every amino acid substitution has an impact of certain magnitude on the alleles function and demonstrate how surrounding residues orchestrate peptide specificity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Defining permissive and non-permissive mismatches for transplantation is a walk on a tightrope. Some mismatches affect the peptide binding motif (Badrinath et al. 2012a; Bade-Doeding et al. 2004, 2007; Elamin et al. 2010), some affect the selection and loading of peptides through differential interaction with the peptide loading complex (Peh et al. 1998; Blanco-Gelaz et al. 2009; Badrinath et al. 2012b), however in some cases, even a single mismatch in the HLA class I locus can tip the balance and trigger an immunological reaction (Fleischhauer et al. 1990; Macdonald et al. 2003, 2009).
The residues within the peptide binding groove (PBR) determine the selection and presentation of antigenic peptides that are surveyed by T cells. These PBR residues contacting the peptide can be subdivided into pockets (Chelvanayagam 1996; Saper et al. 1991) that reflect binding to a certain position of the bound peptide. The choice for C-terminal peptide binding motifs at the pΩ position of the peptide is limited mainly to hydrophobic residues due to the nature of proteasomal processing machinery (Belich and Trowsdale 1995). Subtle alterations in the contacting residues that form the F pocket can either restrict or abrogate the sequence specificity at the pΩ position of the peptide, as we have previously shown for alleles from the HLA-A*74 group (Bade-Doeding et al. 2011b).
Within certain HLA allelic groups, distinct mismatches occur; thus knowledge about the individual peptide binding motif of each individual allele can provide valuable information to predict and understand their impact. The aim of the present study was to identify the unknown peptide binding motif of HLA-B*44:09. The B*44:09 allele represents a special case within the B*44 group, since all the polymorphic residues (residues 77, 80, 81, 82 and 83) are located consecutively within the alpha 1 domain. Residues 77, 80 and 81 comprise part of the F pocket (Chelvanayagam 1996) which determines sequence specificity at the pΩ position of the peptide. Meanwhile residues 82 and 83 lie exposed at the C-terminal end of the peptide binding groove as seen in the structures of B*44 variants (Theodossis et al. 2010) and therefore are not assumed to either bind peptide or influence sequence specificity.
Peptide motif-based ranking of allelic mismatches can contribute to the donor selection process when no HLA-identical donor is available to prevent mismatching being a matter of chance.
Material and methods
Design of lentiviral vectors for sHLA expression
For expression of truncated B*44:09 molecules, a lentiviral vector encoding for exon 1–4 was produced by side-direct mutagenesis using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, Amsterdam, The Netherlands). The pRRL.PPT.SFFV.mcs.pre/B*44:02 vector (Bade-Doeding et al. 2011a) was used as template.
Production of lentiviral particles in HEK293T cells and transduction of B-LCLs
HEK293T cells were transfected with the lentiviral construct pRRL.PPT.SFFV.mcs.pre/B*44:09, the packaging plasmid psPAX2 and the envelope plasmid pDM2G as described previously (Bade-Doeding et al. 2011a).
The B-lymphoblastic cell line LCL 721.221 was then transduced with the lentiviral particles as previously described.
Large-scale production of sHLA molecules and affinity purification
sHLA expression was verified by ELISA as described (Bade-Doeding et al. 2011a). Highly producing clones were cultured and expanded in bioreactors (CELLine, Integra, Fern-wald, Germany). Supernatant containing sHLA-B*44:09 molecules was affinity purified using NHS (N-hydroxysuccimide) activated HiTrap columns, pre-coupled with mab W6/32 (Barnstable et al. 1978; Brodsky et al. 1979). Purification was performed using a BioLogic DuoFlow System (Bio-Rad, Hercules, USA).
Characterization of sHLA-B*44:09-derived peptides
Peptides were eluted from trimeric sHLA_B*44:09 complexes by treatment with 0.1 % trifluoroacetic acid. Peptides were then separated by filtering them through an YM membrane with a 10-kD cutoff (Millipore, Schwalbach, Germany). The flowthrough containing the eluted peptides was then subjected to an Eksigent nano-LC Ultra 2D HPLC coupled to an Orbitrap ion trap (Thermo Fischer, Waltham, Massachusetts, USA). Subsequent database queries were performed using Mascot software (Hirosawa et al. 1993) implementing the IPI human and the respective decoy databases.
Structure analysis and molecular modelling
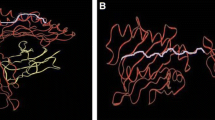
A model of HLA-B*44:09 was generated using DeepView (Guex and Peitsch 1997) with the B*44:02 structure (PDB code 3L3D) (Theodossis et al. 2010) as a template by mutating the HLA heavy-chain residues 77 Asn > Ser, 80 Thr > Asn, 81 Ala > Leu, 82 Leu > Arg and 83 Arg > Gly and the pΩ position of the peptide to W (Fig. 1). Modelling was performed using the internal rotamer library to find the best side chain orientations with minimum steric clashes. Each model was then subjected to energy minimization as implemented in DeepView. The graphics program PyMOL (http://www.pymol.org) was used to generate the structure figures.

Residue 81 is responsible for the peptides C-terminal L/F restriction of B*44:09. a The structure of HLA-B*44:02 (PDB code 3L3D) (Theodossis et al. 2010) illustrated as ribbons with heavy chain coloured blue and bound peptide coloured green. Those residues forming the F pocket are coloured yellow and illustrated as sticks, while the residues 77, 80, 81, 82 and 83 are coloured cyan. b A magnified view illustrating the polymorphic differences between B*44:02 (cyan) and B*44:09 (pink). For B*44:02 the smaller residue 81Ala allows the amino acid W (green) to be accommodated at the p9/pΩ position of the peptide while for B*44:09 the polymorphism 81Leu restricts the pocket size resulting in a preference for smaller hydrophobic residues like F (orange)
Results
Peptide features
We sequenced by LC-ESI-MSMS (LTQ-Orbitrap) technology single peptides derived from sHLA-B*44:09 molecules. Exclusively those peptides were selected for analysis that had a δ value of 0.0 Da and excluded all other peptides. Using these stringent criteria a total of 214 individual peptides (nonduplicates) could be identified (“Electronic supplementary material” (ESM) Table 1). Further analysis of the sequences for the identified peptides illustrated a clear peptide binding motif of E at p2 (150/214 = ~70 %) and predominantly L (100/214 = ~47 %) or F (47/214 = ~22 %) at pΩ (Fig. 2a), with the majority of the identified peptides being 9–11 amino acids in length (Fig. 2c).

Frequency of C-terminal amino acids and frequency of peptide length for B*44:09. Frequency of C-terminal amino acids for HLA-B*44:09. Peptide sequences demonstrated a pΩ anchor motif frequency of L and F (a) while shared peptides between HLA-B*44:02 and B*44:09 predominantly exhibit F at the pΩ position (b). The majority of HLA-B*44:09 selected peptides feature a length of 9–11 amino acids (c)
B*44:09-derived peptides were subsequently compared with the previously described peptides of HLA-B*44:02 (Bade-Doeding et al. 2011a).
Shared peptides
From this and our previous study, we isolated a total of 408 peptides, 214 from sB*44:09 and 194 from sB*44:02 (ESM Table 1 and (Bade-Doeding et al. 2011a)). From this large set of data, we identified 16 shared peptides common to B*44:09 and B*44:02 of which 4 were identical to the 19 previously published peptide sequences for B*44:02 (Hillen et al. 2008) (http://www.syfpeithi.de/). A further seven peptides found only in this study were also common peptides previously identified for B*44:02 and deposited in the syfpeithi database. Thus there are a total of 23 unique shared peptides 16our study + 7syfpethi (Hillen, Lemmel) from 423 total unique peptides 408Our Study + (19Syfpethi − 4 Duplicates(Our study and Syfpethi)) (23/423 = 5.4 %) that we can demonstrate to be shared between B*44:09 and B*44:02 (Table 1). These shared peptides predominantly exhibit F at pΩ (Fig. 2b).
Structural analysis
To investigate the structural basis for the modified C-terminal pΩ anchor observed in peptides bound to B*44:09, we generated a molecular model of B*44:09. The polymorphic residues in the structure of B*44:02 (PDB code 3L3D) (Theodossis et al. 2010) were replaced with those from B*44:09 (see “Material and methods”). In Fig. 1a, the structure of B*44:02 is illustrated and those residues forming the F pocket are coloured yellow, while the residues 77, 80, 81, 82 and 83 that are polymorphic between B*44:02 and B*44:09 are coloured cyan. Residues 77, 80 and 81 are known to be part of pocket F and contact the pΩ anchor position of bound peptides, while residues 82 and 83 lie on the periphery of the peptide binding groove and play no role in peptide binding. In Fig. 1b, the structures of B*44:02 and our B*44:09 model are superimposed. From this close-up view, it is clear that residues 77 and 80 make contact with the peptide main chain at the pΩ position, while 81 contacts the pΩ side chain. Residue 81Leu (B*44:09) appears to be the main cause of pΩ sequence specificity by restricting the size of the F pocket, resulting its in selection against a bulky residue such as W at this position (Fig. 1b).
Discussion
The functional differences of HLA molecules are reflected by their ability to select and present a broad spectrum of peptides. Each HLA allele has an individual peptide binding motif that specifies the preferred and auxiliary anchor residues which is derived from an analysis of the naturally presented peptide repertoire (selection of all available ligands).
We determined the bound peptide features of HLA-B*44:09 and analyzed the impact of the residues polymorphic distinguishing B*44:09 from B*44:02. B*44:09 differs from the highly frequent B*44:02 allele by five polymorphisms (77 Asn > Ser, 80 Thr > Asn, 81 Ala > Leu, 82 Leu > Arg and 83 Arg > Gly). From these residues 77, 80 and 81 comprise part of the F pocket (Chelvanayagam 1996) which determines sequence specificity at the pΩ position of the peptide, while residues 82 and 83 do not either bind peptide or influence sequence specificity. We found B*44:09 to partially share the peptide binding motif and certain single peptides with the common B*44:02 despite the described polymorphism. The peptide binding motifs of B*44:09 and B*44:02 are similar in that they contain an E anchor at p2 and both alleles accommodate F and L at pΩ of the peptide. However, despite the alleles demonstrating binding motif similarities and even sharing a partial peptide repertoire, B*44:02 specifically selects peptides containing W at the pΩ position by virtue of its larger F pocket. The restriction in the size of the F pocket for B*44:09 is caused primarily by the Ala81 > Leu polymorphism as illustrated by structural modelling (Fig. 1). The key question however is, will the alteration in the pHLA landscape arouse CTL responses in both transplantation directions? In this respect, the differences and composition of the shared peptides between the two alleles suggest that permissivity might be considered unidirectional from B*44:09 to B*44:02 since B*44:02 can present peptides with the B*44:09 peptide binding motif and not vice versa.
Peptide sharing and promiscuity among HLA class I molecules is not novel. Indeed, peptide sharing between different loci has previously been described for certain predicted viral epitopes (Frahm et al. 2007; Rao et al. 2011). However, the polymorphic differences between the B*44:02 and B*44:09 alleles result in an altered F pocket, thus we had not expected any significant overlap in the peptide anchor motifs for these alleles. Our observation of 5 % shared peptides highlights that those mismatches discriminating B*44:02 and B*44:09 still result in the selection of a small overlapping repertoire, at least in the context of those peptides processed and presented by the B-LCL 721.221 cells. Although this does not provide sufficient evidence that both alleles can share specific viral or immunogenic epitopes, this possibility can also not been ruled out. This study provides new knowledge about HLA class I allelic promiscuity for naturally processed and presented peptides and is of fundamental interest in the quest to understand T cell allorecognition, viral epitope expression kinetics, processing and presentation. The work presented here also continues our efforts to understand and predict HLA mismatching through biochemical and structural analysis of specific alleles. Thus, by extending our knowledge of how polymorphism influences the magnitude of HLA mismatching we can make better decisions in choosing donors, when no complete match is available.
References
Bade-Doeding C, Cano P, Huyton T, Badrinath S, Eiz-Vesper B, Hiller O, Blasczyk R (2011a) Mismatches outside exons 2 and 3 do not alter the peptide motif of the allele group B*44:02P. Hum Immunol 72:1039–1044
Bade-Doeding C, Huyton T, Eiz-Vesper B, Blasczyk R (2011b) The composition of the F pocket in HLA-A*74 generates C-terminal promiscuity among its bound peptides. Tissue Antigens 78:378–81
Bade-Doeding C, DeLuca DS, Seltsam A, Blasczyk R, Eiz-Vesper B (2007) Amino acid 95 causes strong alteration of peptide position Pomega in HLA-B*41 variants. Immunogenetics 59:253–259
Bade-Doeding C, Elsner HA, Eiz-Vesper B, Seltsam A, Holtkamp U, Blasczyk R (2004) A single amino-acid polymorphism in pocket A of HLA-A*6602 alters the auxiliary anchors compared with HLA-A*6601 ligands. Immunogenetics 56:83–88
Badrinath S, Huyton T, Schumacher H, Blasczyk R, Bade-Doeding C (2012a) Position 45 influences the peptide binding motif of HLA-B*44:08. Immunogenetics 64:245–9
Badrinath S, Saunders P, Huyton T, Aufderbeck S, Hiller O, Blasczyk R, Bade-Doeding C (2012b) Position 156 influences the peptide repertoire and tapasin dependency of human leukocyte antigen B*44 allotypes. Haematologica 97:98–106
Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, Ziegler A (1978) Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 14:9–20
Belich MP, Trowsdale J (1995) Proteasome and class I antigen processing and presentation. Mol Biol Rep 21:53–6
Blanco-Gelaz MA, Suarez-Alvarez B, Diaz-Pena R, Lopez-Larrea C (2009) HLA-B27 polymorphism at position 116 critically influences the association with TAP/tapasin, intracellular trafficking and conformational homodimers formation. Mol Immunol 46:1304–11
Brodsky FM, Parham P, Barnstable CJ, Crumpton MJ, Bodmer WF (1979) Monoclonal antibodies for analysis of the HLA system. Immunol Rev 47:3–61
Chelvanayagam G (1996) A roadmap for HLA-A, HLA-B, and HLA-C peptide binding specificities. Immunogenetics 45:15–26
Elamin NE, Bade-Doeding C, Blasczyk R, Eiz-Vesper B (2010) Polymorphism between HLA-A*0301 and A*0302 located outside the pocket F alters the POmega peptide motif. Tissue Antigens 76:487–90
Fleischhauer K, Kernan NA, O'Reilly RJ, Dupont B, Yang SY (1990) Bone marrow-allograft rejection by T lymphocytes recognizing a single amino acid difference in HLA-B44. N Engl J Med 323:1818–22
Frahm N, Yusim K, Suscovich TJ, Adams S, Sidney J, Hraber P, Hewitt HS, Linde CH, Kavanagh DG, Woodberry T, Henry LM, Faircloth K, Listgarten J, Kadie C, Jojic N, Sango K, Brown NV, Pae E, Zaman MT, Bihl F, Khatri A, John M, Mallal S, Marincola FM, Walker BD, Sette A, Heckerman D, Korber BT, Brander C (2007) Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur J Immunol 37:2419–33
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714–23
Hillen N, Mester G, Lemmel C, Weinzierl AO, Muller M, Wernet D, Hennenlotter J, Stenzl A, Rammensee HG, Stevanovic S (2008) Essential differences in ligand presentation and T cell epitope recognition among HLA molecules of the HLA-B44 supertype. Eur J Immunol 38:2993–3003
Hirosawa M, Hoshida M, Ishikawa M, Toya T (1993) MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming. Comput Appl Biosci 9:161–7
Lemmel C, Weik S, Eberle U, Dengjel J, Kratt T, Becker HD, Rammensee HG, Stevanovic S (2004) Differential quantitative analysis of MHC ligands by mass spectrometry using stable isotope labeling. Nat Biotechnol 22:450–4
Macdonald WA, Chen Z, Gras S, Archbold JK, Tynan FE, Clements CS, Bharadwaj M, Kjer-Nielsen L, Saunders PM, Wilce MC, Crawford F, Stadinsky B, Jackson D, Brooks AG, Purcell AW, Kappler JW, Burrows SR, Rossjohn J, McCluskey J (2009) T cell allorecognition via molecular mimicry. Immunity 31:897–908
Macdonald WA, Purcell AW, Mifsud NA, Ely LK, Williams DS, Chang L, Gorman JJ, Clements CS, Kjer-Nielsen L, Koelle DM, Burrows SR, Tait BD, Holdsworth R, Brooks AG, Lovrecz GO, Lu L, Rossjohn J, McCluskey J (2003) A naturally selected dimorphism within the HLA-B44 supertype alters class I structure, peptide repertoire, and T cell recognition. J Exp Med 198:679–91
Peh CA, Burrows SR, Barnden M, Khanna R, Cresswell P, Moss DJ, McCluskey J (1998) HLA-B27-restricted antigen presentation in the absence of tapasin reveals polymorphism in mechanisms of HLA class I peptide loading. Immunity 8:531–42
Rao X, Hoof I, Costa AI, van Baarle D, Kesmir C (2011) HLA class I allele promiscuity revisited. Immunogenetics 63:691–701
Saper MA, Bjorkman PJ, Wiley DC (1991) Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J Mol Biol 219:277–319
Theodossis A, Guillonneau C, Welland A, Ely LK, Clements CS, Williamson NA, Webb AI, Wilce JA, Mulder RJ, Dunstone MA, Doherty PC, McCluskey J, Purcell AW, Turner SJ, Rossjohn J (2010) Constraints within major histocompatibility complex class I restricted peptides: presentation and consequences for T-cell recognition. Proc Natl Acad Sci U S A 107:5534–9
Acknowledgments
The technical assistance of Susanne Aufderbeck is gratefully acknowledged. This work was supported by a grant from the German Federal Ministry of Education and Research (reference number 01E00802).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM 1
(DOC 77 kb)
Rights and permissions
About this article
Cite this article
Huyton, T., Schumacher, H., Blasczyk, R. et al. Residue 81 confers a restricted C-terminal peptide binding motif in HLA-B*44:09. Immunogenetics 64, 663–668 (2012). https://doi.org/10.1007/s00251-012-0625-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-012-0625-1