
Abstract
The largest remaining carnivorous marsupial, the Tasmanian devil (Sarcophilus harrisii), is currently under threat of extinction due to a fatal contagious cancer—devil facial tumour disease. Low major histocompatibility complex (MHC) class I diversity is believed to have contributed to the transmission of the tumour allograft through devil populations. Here, we report low MHC class II variability in this species, with DA β chain genes (Saha-DAB1, 2 and 3) exhibiting very limited diversity and the sole α chain gene (Saha-DAA) monomorphic. Three, six and three alleles were found at Saha-DAB1, 2 and 3, respectively, with a predominant allele found at each locus. Heterozygosity at these three loci is low in the eastern population and modestly higher in northwestern individuals. The results are indicative of a selective sweep likely due to an infectious disease resulting in the fixation of selectively favoured alleles and depletion of genetic diversity at devil class II loci. Several attempts were made to isolate the other marsupial classical class II gene family, namely, DB, resulting in only one DBB pseudogene being found. These findings further support the view that this species has a compromised capacity to respond to pathogen evolution, emerging infectious diseases and environmental changes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) is a gene-rich region found in all jawed vertebrate genomes which plays a significant role in disease resistance and transplantation success (Janeway et al. 2001). MHC class II genes are centrally involved in the efficient production of antibody responses and successful defence against bacterial and parasitic infections (Frank 2002). Class II molecules are normally expressed on the surface of immune cells, including B lymphocytes, dendritic cells and macrophages. These molecules function by binding peptides derived from intravesicular and extracellular pathogens and presenting these antigens to CD4+ helper T cells (reviewed in Rocha and Neefjes 2008). An MHC class II molecule is a heterodimer of two non-covalently associated polypeptide chains, an α chain and a β chain, both of which consist of a peptide-binding domain (α1 and β1 domains), an immunoglobulin-like domain (α2 and β2 domains) and a membrane-spanning domain (Brown et al. 1993). As a result of host–pathogen co-evolution, high genetic polymorphism is usually found in the peptide-binding region within the α1 and β1 domains, which enables class II molecules to bind a diverse set of peptides and thereby allows the immune system to respond to an extensive range of pathogens (O’Brien and Evermann 1988). In wildlife conservation studies, the level of MHC class II variability represents an important indicator of the capacity of wild populations to mount immune responses to a range of infectious diseases (Edwards and Potts 1996).
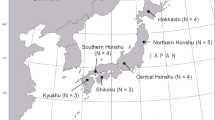
The Tasmanian devil (Sarcophilus harrisii; ‘devil’ for short) is an endangered carnivorous marsupial that is endemic in the island of Tasmania, Australia. This island population has been isolated for over 12,000 years, and as a result of island effects and historical bottlenecks, it suffers from a low level of genetic diversity (Jones et al. 2004; Miller et al. 2011). In recent years, the devil has undergone rapid population declines due to the emergence of devil facial tumour disease (DFTD), which is caused by a contagious tumour cell line that appears to have originated from a Schwann cell and is able to be transmitted between unrelated individuals as an allograft by cellular inoculation (e.g. biting) (Pearse and Swift 2006; Murchison et al. 2010). Since first detection in 1996 in northeastern Tasmania, DFTD has spread to over 85 % of the original devil range, leaving only a small region in western Tasmania currently unaffected (Fig. 1; Hamede et al. 2008). According to the 2010/11 Annual Report of the Save the Tasmanian Devil Program (available at http://www.tassiedevil.com.au), DFTD has resulted in an 84 % decline in population size, which was estimated to range up to 150,000 prior to 1996 (Hawkins et al. 2006). The severe population decline has led to significant changes in genetic structure and dispersal patterns of the species, and it is predicted that the disease may eventually cause extinction of the devil in the wild (McCallum et al. 2009; Lachish et al. 2011).

Map of Tasmania showing the site of first detection of DFTD, the current location of disease front and the sampling sites
The level of genetic diversity at devil MHC class I genes has been examined previously (Siddle et al. 2010). Extremely low levels of class I diversity were found in the east, whereas slightly higher levels of diversity as well as gene copy number variation were found in the northwest. Here, we report low MHC class II genetic diversity in devil populations. Marsupials typically have two classical class II gene families, designated DA and DB (Belov et al. 2006; Siddle et al. 2011). A single DA α chain gene (Saha-DAA) and three DA β chain genes (Saha-DAB1, 2 and 3) have been identified in the devil genome by screening a devil bacterial artificial chromosome (BAC) library with 10× genome coverage (Cheng et al. 2012). Here, we examine the genetic variation at these four loci. We also propose that DA is the only functional class II gene family in the Tasmanian devil.
Materials and methods
Tasmanian devil samples
Sixty individuals were used in this study, 30 from eastern population and 30 from DFTD-free northwestern Tasmania (Fig. 1). Genomic DNA of all 60 individuals was extracted from fresh or frozen (stored at −20°C) blood or from ear biopsies using DNeasy Blood and Tissue Kit (QIAGEN).
Assessment of genetic diversity at four DA loci
DA α chain gene Saha-DAA
Polymerase chain reaction (PCR) primers (forward 5′-CATCCAAGCTGAGTTCTACC-3′ and reverse 5′-TTGTTGGACCGTTTTATCAT-3′) amplifying a 216-bp fragment from exon 2 (Fig. 2; GenBank, FQ790241:10943-10728) were designed based on Saha-DAA sequence extracted from the BAC sequence (Cheng et al. 2012). Primer design was carried out using programme Oligo 6.7 (Molecular Biology Insights). PCRs were performed in a total volume of 25 μL containing 1× high-fidelity buffer (Invitrogen) that consists of 60 mM Tris–HCl (pH 8.9) and 18 mM (NH4)2SO4, 2.0 mM MgSO4, 0.2 mM each dNTP, 0.5 μM each primer, 1.5 U of Platinum Taq DNA Polymerase High Fidelity (Invitrogen) and approximately 40 ng template DNA. High-fidelity Taq was used to ensure lowest error rate. The PCRs were carried out on a Bio-Rad MJ Mini Personal Thermal Cycler at the following conditions: 100°C hot lid; 94°C initial denaturation for 3 min; 33 cycles of 94°C denaturation for 30 s, 60°C annealing for 30 s, and 72°C extension for 30 s; and 72°C final extension for 10 min. PCR amplicons were isolated by running a 1.8 % agarose gel using HyperLadder IV (Bioline) as size marker and purified from the gel using QIAquick Gel Extraction Kit (QIAGEN). The DNA fragments were then cloned in a pGEM-T Easy Vector/JM109 High Efficiency Competent Cells (Promega) cloning system. Four positive clones were picked for each individual and plasmids were extracted using QIAprep Spin Miniprep Kit (QIAGEN) or DirectPrep 96 MiniPrep Kit (QIAGEN) on a QIAvac Multiwell vacuum manifold (QIAGEN). Plasmids were sequenced with T7 primer at the Australian Genome Research Facility, Sydney, Australia.

Amino acid alignment of partial exon 2 of Tasmanian devil DAA allele with red-necked wallaby (Maru) and opossum (Modo) DAA and human DRA sequences. The ruler has been adjusted according to the human DRA sequence
DA β chain gene Saha-DAB1, 2 and 3
A single pair of primers (forward 5′-GGTCCCCGCAGAGCACTTCAC-3′ and reverse 5′-GCCTGCGCACTAAGAAGGACTC-3′) was used to amplify a 278-bp fragment of β1 domain (exon 2) from the three DAB loci (Cheng et al. 2012). Protocols for PCR amplification, PCR product cloning, plasmid purification and sequencing were same as above, except that two independent PCRs were carried out for each individual and 10–16 clones were sequenced for each PCR sample.
Sequence analysis
Sequences were quality-checked using Sequencher 4.1.4 (Gene Codes) and aligned with previous isolated devil class II sequences (Siddle et al. 2007) in BioEdit 7.0.9 (Hall 1999). To minimise errors from PCR, cloning and sequencing, new sequence variants were determined to be real alleles only if they were found in more than one PCR amplification. We estimated that the sequencing error rate was approximately 0.6 % and the frequency of occurrence of artificial chimeras was lower than 2 %. Phylogenetic analysis was conducted in MEGA4 (Tamura et al. 2007) using the neighbour-joining method (Saitou and Nei 1987) with 1,000 bootstrap replicates to infer the level of confidence on the phylogeny (Felsenstein 1985). Analysis of synonymous and nonsynonymous nucleotide substitutions within and outside the peptide-binding region (PBR) was computed in MEGA4 using the modified Nei–Gojobori method with Jukes–Cantor correction to account for multiple substitutions at a single site (Nei and Kumar 2000). Five thousand bootstrap replications were used to generate the standard error for Z test. For each DAB locus, the allele frequency and observed and expected heterozygosity were calculated and tested for Hardy–Weinberg equilibrium using Cervus 3.0 (Kalinowski et al. 2007). The level of population differentiation between eastern and northwestern devils was estimated in FSTAT 2.9.3.2 (Goudet 2001).
The following class II sequences from the GenBank database were used in phylogenetic analysis: tammar wallaby (Macropus eugenii)—MaeuDAB01 (AY438042), MaeuDAB02-05 (AY856411-4) and MaeuDBB*01 (AY438038); red-necked wallaby (Macropus rufogriseus)—MaruDBB (M81625) and MaruDAA (U18109); brushtail possum (Trichosurus vulpecula)—TrvuDBB*0101-3 (EU500907-9), TrvuDAB*0101-3 (EU500877-9), TrvuDAA*0101 (EU500871), TrvuDAA*0102 (EU500874), TrvuDBA*01 (EU500895) and TrvuDBA*02 (EU500901); grey short-tailed opossum (Monodelphis domestica)—ModoDAB (AF010497), ModoDAA, ModoDBA*1, ModoDBA*2, ModoDBB*1 and ModoDBB*2 are available at http://bioinf.wehi.edu.au/opossum/seq/Class_II.fa; cattle (Bos taurus)—BotaDRB (NM_001012680), BotaDQB (NM_001034668), BotaDRA (NM_001012677) and BotaDQA (NM_001013601); human—HLA-DPA1 (NM_033554), HLA-DPB1 (NM_002121), HLA-DQA1 (NM_002122), HLA-DQB1 (NM_002123), HLA-DRA (NM_019111) and HLA-DRB1 (NM_002124); and African clawed frog (Xenopus laevis)—XelaDRA (NM_001094869) and XelaDRB (NM_001114771).
Searching for DB loci
Several approaches were employed to identify DB family loci in the devil. First, a cDNA library was constructed and screened. Total RNA was extracted from fresh whole blood of a northwestern devil using RNeasy Mini Kit (QIAGEN). A cDNA library with a titer of ∼8.8 × 106 pfu/ml before amplification was produced using SMART cDNA Library Construction Kit (Clontech). Full-length cDNA was ligated into λTriplEx2 Vector followed by packaging into λ phage with Gigapack III Gold Packaging Extract (Stratagene). A tammar wallaby DBB fragment was PCR amplified (Cheng et al. 2009) and used as probe to screen the cDNA library following previously described protocols (Belov et al. 2003).
Second, the devil genome on Ensembl (available at http://www.ensembl.org/Sarcophilus_harrisii) was BLAST searched using the grey short-tailed opossum DBA and DBB sequences (available at http://bioinf.wehi.edu.au/opossum/seq/Class_II.fa). Only one DBB β2-like segment (∼219 bp) was identified, for which PCR primers were designed: forward 5′-CTAAACAAGATCAAGGTCAC-3′ and reverse 5′-ATGACAAGGGTCTGGTAGGT-3′. PCR protocol used to amplify this fragment was same as the one described above.
Third, this DBB β2-like fragment was used as a probe to screen the devil BAC library VMRC-49 (Cheng et al. 2012). One positive clone containing the fragment was identified and sequenced at the Wellcome Trust Sanger Institute, Cambridge, UK.
Results
Genetic variation at Saha-DAA
Only one Saha-DAA allele, SahaDAA*01, was found in all 60 examined animals, which is identical to the one identified in the BAC sequence (Fig. 2; GenBank, FQ790241:10943-10728). The phylogenetic relationship of this sequence and MHC class II α chain genes from other species is shown in Fig. 3. Saha-DAA forms a clade with other marsupial DAA genes with 100 % bootstrap support, showing a closer evolutionary relationship with orthologues from other Australian marsupials (red-necked wallaby and brushtail possum) than with the South American grey short-tailed opossum DAA.

Phylogenetic analysis of MHC class II α and β chain sequences. The phylogenetic relationship was inferred using the neighbour-joining method with 1,000 bootstrap replicates. Bootstrap frequencies lower than 40 % are not shown
Genetic variation at Saha-DAB1, 2 and 3 in two populations
Thirty eastern and 30 northwestern devils were examined at DAB loci. A total of 12 DAB alleles were identified, three of which (SahaDAB*01, 03 and 05) have been reported previously and represent the three DAB genes (Saha-DAB1, 2 and 3), respectively (Siddle et al. 2007; Cheng et al. 2012). Interestingly, every single animal typed has at least one of each of these alleles. Nine new alleles were named as SahaDAB*07-15 (GenBank, JQ065645-JQ065653) in consistency with formerly used nomenclature for devil DAB alleles. Close phylogenetic relationships were detected between alleles SahaDAB*12, 13 and 01, SahaDAB*08, 09, 10, 14, 15 and 03 and SahaDAB*07, 11 and 05 with high bootstrap confidence levels, inferring the likely assignment of the novel alleles to the three loci (Fig. 3). Three to five alleles, corresponding to one to two alleles from each locus, were found in each individual (Supplementary material S1). All devil DAB alleles, together with other marsupial DAB genes, fall into a clade that is separated from the marsupial DBB, eutherian DRB and DQB and amphibian DRB with 98 % bootstrap support. Within this marsupial DAB clade, sequences from the devil, tammar wallaby and brushtail possum are well separated by species, indicating that gene duplication events that gave rise to the three devil DAB genes occurred after the divergence of these three marsupial lineages at ∼66 million years before present (Kirsch et al. 1997).
Low polymorphism levels were found at each devil DAB locus, with the number of alleles ranging from three to six and the number of amino acid variations between alleles less than five (Fig. 4). SahaDAB*01, 03 and 05 are the predominant Saha-DAB1, 2 and 3 alleles, respectively, in both eastern and northwestern populations, with respective allele frequencies of 0.850, 0.825 and 0.900 (Fig. 5). No significant deviations from Hardy–Weinberg equilibrium were detected, suggesting that the two populations are undergoing random mating. Eastern and northwestern devils share most DAB alleles except SahaDAB*07, which was only identified in northwestern individuals. Based on the estimates of fixation index and exact tests of population subdivision (Weir and Cockerham 1984; Goudet et al. 1996), the genetic differentiation at Saha-DAB1, 2 and 3 is weak between the two populations, with F ST being −0.015 (p = 0.99), 0.004 (p = 0.81) and 0.003 (p = 0.12), respectively. However, consistent with what have been found in class I genes (Siddle et al. 2010), relatively higher heterozygosity values were observed in the northwestern individuals (0.333, 0.433 and 0.233) than in the east (0.300, 0.267 and 0.167) at all three DAB loci (Table 1).

Amino acid alignment of partial exon 2 of 12 Tasmanian devil DAB alleles. Alleles from each of the three DAB loci are aligned separately. To facilitate analysis, the ruler has been adjusted according to the human DRB sequence. Asterisks indicate putative peptide-binding sites (Bondinas et al. 2007)

Frequency of 12 Tasmanian devil DAB alleles in eastern and northwestern populations
By comparing synonymous and nonsynonymous nucleotide substitution rates at amino acid sites within and outside the PBR, we detected signs of positive selection at devil DAB loci (Table 2). Within the PBR of Saha-DAB2 and 3, the mean number of nonsynonymous substitutions per nonsynonymous site (d N) is significantly higher (at 0.05 nominal level) than the mean number of synonymous substitutions per synonymous site (d S), demonstrating that the PBRs of these two loci are targets of balancing selection (Hughes 1999; Nei and Kumar 2000).
DBB pseudogene
No DB transcripts were isolated from the cDNA library. BLAST searches of the devil genome revealed one DBB β2 fragment. The DBB fragment was used to isolate a BAC clone, which was then sequenced to reveal a DBB pseudogene (GenBank, FQ790240:26037-19222) that lacks intact exons 2 and 3. PCR confirmed that this pseudogene is not transcribed in blood, spleen or DFTD tumour.
Discussion
High MHC class II diversity plays a crucial role in the host defence against a variety of infectious diseases and is primarily ensured by two compensatory mechanisms. First, gene loss within one gene family is usually accompanied by gene expansion in another one. One example is the domestic cat (Felis catus). Compared to the human HLA, which contains three sets of classical class II genes, DR, DQ and DP, the cat MHC lacks the entire DQ gene family and retains two DP psaeudogenes (Yuhki et al. 2003). Nevertheless, high class II variability is achieved in the cat via species-specific expansion of seven modern DR genes, comprising three α chain genes and four β chain genes. Second, limited polymorphism in one subunit of the class II heterodimer is commonly compensated by high variation in the other one. For instance, although low DRA diversity is seen in humans, cats, dogs and pigs (Chu et al. 1994; Yuhki and O’Brien 1997; Wagner et al. 1999; Ho et al. 2009), the DRB genes of these species are highly diverse, contributing to an overall high variability of DR molecules. A similar situation occurs in the brushtail possum with high polymorphism at DAB loci making up for low DAA diversity (Holland et al. 2008a, b).
Neither of these two compensatory mechanisms has been detected in the Tasmanian devil MHC, resulting in an unusually low level of class II diversity in this species. The devil appears to have only one functional class II gene family—DA. Multiple functional class II DB genes have been identified in the grey short-tailed opossum, tammar wallaby and brushtail possum. A sole DBB pseudogene remains in the devil genome, suggesting that this gene family was long lost in this species. Despite this gene family loss, no expansion is seen in the other class II gene family. Four DA genes have been identified in the devil, which is comparable to the number estimated in the brushtail possum and the Brazilian gracile mouse opossum (Gracilinanus microtarsus), and much less than that found in the tammar wallaby (Table 3). Genetic polymorphism in the four devil DA genes is highly limited. The sole α chain gene identified in the devil, Saha-DAA, is monomorphic. At DAB loci, not only do the three genes share high exon sequence similarity (>95.3 %) (Cheng et al. 2012), very few single nucleotide polymorphisms are found at each locus. The reduced allelic richness and high frequency of the predominant allele lead to low heterozygosities at these β chain loci. In fact, 35 % of examined individuals was homozygous at all three loci, which means that these devils only possess a set of three highly similar class II molecules (Supplementary material S1). Such low levels of class II diversity are rarely observed in marsupials (Table 3), even in long-term isolated island populations such as the Kangaroo Island tammar wallaby (Cheng et al. 2009) or bottlenecked populations such as the introduced New Zealand brushtail possum (Holland et al. 2008a, b), both of which show high class II diversity. In the grey slender mouse opossum (Marmosops incanus), which is endemic to South America, despite the small number of alleles found at DAB loci, high sequence divergence is seen between the alleles (Meyer-Lucht et al. 2008).
The low class II diversity in the devil is likely attributed to both genetic drift on the island population plus founder effects after population bottlenecks. Purifying selection may have acted specifically on the MHC loci, resulting in a selective sweep for the predominant, presumably favourably ‘fit’ MHC alleles, possibly due to infectious disease. Low genetic variation is also seen at devil class I genes (Siddle et al. 2010), although the allelic richness and sequence divergence are higher at class I loci compared to class II. A similar process appears to have occurred in chimpanzees where a selective sweep, which may have been caused by a simian immunodeficiency virus and primarily targeted MHC class I genes, resulted in a severe reduction of allelic repertoire at these loci and also affected other polymorphic loci near the class I region due to genetic linkage (de Groot et al. 2002, 2008). In the devil, it is hard to pinpoint the direct target of the selective sweep as class I and class II genes are tightly linked (Cheng et al. 2012), which means the sweep could have taken place in either region and affected the other through linkage. It has been suggested that the Tasmanian devil experienced a population crash in the late nineteenth to early twentieth century due to an epidemic disease, which also led to the extinction of the thylacine (Thylacinus cynocephalis, Tasmanian tiger) (Paddle 2000, 2012). This disease was possibly viral and was described as distemper-like, though the aetiological agent is unknown (Paddle 2000, 2011). Reports from captive devils showed that some individuals survived repeated contact with the disease while others were highly susceptible (Paddle 2012), providing support for the opportunity of strong selection on the MHC. Strong purifying selection could also have been caused by emerging diseases from invasive species after European settlement, including toxoplasmosis from feral cats (Beveridge and Spratt 2003).
In conclusion, Tasmanian devils have low levels of genetic diversity at their MHC class II genes. The DA α chain gene is invariable and the β chain genes show very limited polymorphism. DB genes are likely to have been lost in the devil genome. A direct implication of such reduced class II variability is increased vulnerability of the species to evolving pathogens, emerging infectious diseases and environmental changes. Devils are exposed to high levels of microbial and parasitic pathogens, including several Mycobacterium species and Salmonella serotypes (Holz 2008), a range of trematode, cestode and nematode parasites as well as various external parasites (Spratt et al. 1991; Beveridge and Spratt 2003). It is critical for the long-term survival of this species to ensure that further selective sweeps do not occur and that existing MHC allelic diversity is retained.
References
Belov K, Lam MKP, Hellman L, Colgan DJ (2003) Evolution of the major histocompatibility complex: isolation of class II beta cDNAs from two monotremes, the platypus and the short-beaked echidna. Immunogenetics 55:402–411
Belov K, Deakin JE, Papenfuss AT et al (2006) Reconstructing an ancestral mammalian immune supercomplex from a marsupial major histocompatibility complex. PLoS Biol 4:e46
Beveridge I, Spratt DM (2003) Parasites of carnivorous marsupials. In: Jones M, Dickman CR, Archer M (eds) Predators with pouches: the biology of carnivorous marsupials. CSIRO, Melbourne
Bondinas GP, Moustakas AK, Papadopoulos GK (2007) The spectrum of HLA-DQ and HLA-DR alleles, 2006: a listing correlating sequence and structure with function. Immunogenetics 59:539–553
Brown JH, Jardetzky TS, Gorga JC et al (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 364:33–39
Cheng Y, Siddle HV, Beck S, Eldridge MDB, Belov K (2009) High levels of genetic variation at MHC class II DBB loci in the tammar wallaby (Macropus eugenii). Immunogenetics 61:111–118
Cheng Y, Stuart A, Morris K et al. (2012) BAC-contig-based characterization of MHC Class I and Class II regions in the Tasmanian devil (Sarcophilus harrisii). BMC Genom 13:87
Chu Z-TE, Carswell-Crumpton C, Cole BC, Jones PP (1994) The minimal polymorphism of class II Eα chains is not due to the functional neutrality of mutations. Immunogenetics 40:9–20
de Groot NG, Otting N, Doxiadis GGM et al (2002) Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc Natl Acad Sci 99:11748–11753
de Groot NG, Heijmans CMC, de Groot N et al (2008) Pinpointing a selective sweep to the chimpanzee MHC class I region by comparative genomics. Mol Ecol 17:2074–2088
Edwards SV, Potts WK (1996) Polymorphism of genes in the major histocompatibility complex: implications for conservation genetics of vertebrates. In: Smith TB, Wayne RK (eds) Molecular genetic approaches in conservation. Oxford University Press, Oxford
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Frank SA (2002) Immunology and evolution of infectious disease. Princeton University Press, Princeton
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ch/izea/softwares/fstat.html. Updated from Goudet (1995)
Goudet J, Raymond M, de Meeüs T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Hall T (1999) Bioedit: a user friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hamede R, McCallum H, Jones ME (2008) Seasonal, demographic and density-related patterns of contact between Tasmanian devils: implications for transmission of devil facial tumour disease. Austral Ecol 33:614–622
Hawkins CE, Baars C, Hesterman H et al (2006) Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol Conserv 131:307–324
Ho CS, Lunney JK, Ando A et al (2009) Nomenclature for factors of the SLA system, update 2008. Tissue Antigens 73:307–315
Holland OJ, Cowan PE, Gleeson DM, Chamley LW (2008a) High variability in the MHC class II DA beta chain of the brushtail possum (Trichosurus vulpecula). Immunogenetics 60:775–781
Holland OJ, Cowan PE, Gleeson DM, Chamley LW (2008b) Novel alleles in classical major histocompatibility complex class II loci of the brushtail possum (Trichosurus vulpecula). Immunogenetics 60:449–460
Holz PH (2008) Dasyurids. In: Vogelnest E, Woods R (eds) Medicine of Australian mammals. CSIRO, Melbourne
Hughes AL (1999) Adaptive evolution of genes and genomes. Oxford University Press, New York
Janeway CA, Travers P, Walport M, Shlomchik M (2001) Immunobiology: the immune system in health and disease. Garland, New York
Jones ME, Paetkau D, Geffen E, Moritz C (2004) Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol Ecol 13:2197–2209
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Kirsch JAW, Lapointe F-J, Springer MS (1997) DNA-hybridisation studies of marsupials and their implications for Metatherian classification. Aust J Zool 45:211–280
Lachish S, Miller KJ, Storfer A, Goldizen AW, Jones ME (2011) Evidence that disease-induced population decline changes genetic structure and alters dispersal patterns in the Tasmanian devil. Heredity 106:172–182
McCallum H, Jones M, Hawkins C et al (2009) Transmisson dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology 90:3379–3392
Meyer-Lucht Y, Otten C, Puttker T, Sommer S (2008) Selection, diversity and evolutionary patterns of the MHC class II DAB in free-ranging Neotropical marsupials. BMC Genet 9:39
Miller W, Hayes VM, Ratan A et al (2011) Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc Natl Acad Sci 108:12348–12353
Murchison EP, Tovar C, Hsu A et al (2010) The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science 327:84–87
Nei M, Kumar S (2000) Synonymous and nonsynonymous nucleotide substitutions. In: Molecular evolution and phylogenetics. Oxford University Press, New York
O’Brien S, Evermann J (1988) Interactive influence of infectious disease and genetic diversity in natural populations. Trends Ecol Evol 3:254–259
Paddle R (2000) The last Tasmanian tiger: the history and extinction of the Thylacine. Cambridge University Press, Melbourne
Paddle R (2012) The thylacine’s last straw: epidemic disease in a recent mammalian extinction. Aust Zool, in press
Pearse AM, Swift K (2006) Allograft theory: transmission of devil facial-tumour disease. Nature 439:549
Rocha N, Neefjes J (2008) MHC class II molecules on the move for successful antigen presentation. EMBO J 27:1–5
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Siddle HV, Sanderson C, Belov K (2007) Characterization of major histocompatibility complex class I and class II genes from the Tasmanian devil (Sarcophilus harrisii). Immunogenetics 59:753–760
Siddle HV, Marzec J, Cheng Y, Jones M, Belov K (2010) MHC gene copy number variation in Tasmanian devils: implications for the spread of a contagious cancer. Proc R Soc B 277:2001–2006
Siddle H, Deakin J, Coggill P et al (2011) The tammar wallaby major histocompatibility complex shows evidence of past genomic instability. BMC Genomics 12:421
Spratt DM, Beveridge I, Walter EL (1991) A catalogue of Australasian monotremes and marsupials and their recorded helminth parasites. In: Jones PG, Horton P, Mathews EG, Thurmer J (eds) Records of the South Australian Museum. Monograph series 1. South Australian Museum, Adelaide
Stone WH, Bruun DA, Fuqua C et al (1999) Identification and sequence analysis of an Mhc class II B gene in a marsupial (Monodelphis domestica). Immunogenetics 49:461–463
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. doi:10.1093/molbev/msm1092
Wagner J, Burnett R, Storb R (1999) Organization of the canine major histocompatibility complex: current perspectives. J Hered 90:35–38
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Yuhki N, O’Brien S (1997) Nature and origin of polymorphism in feline MHC class II DRA and DRB genes. J Immunol 158:2822–2833
Yuhki N, Beck T, Stephens RM, Nishigaki Y, Newmann K, O’Brien SJ (2003) Comparative genome organization of human, murine, and feline MHC class II region. Genome Res 13:1169–1179
Acknowledgments
This work was funded by the Australian Research Council. KB and MJ are supported by ARC Future Fellowships. YC is supported by an Endeavour International Postgraduate Research Scholarship. We thank Rodrigo Hamede and Shelley Lachish at the University of Tasmania for sample collection.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
S1
Genotype data of 60 Tasmanian devils at MHC Class II loci (PDF 43 kb)
Rights and permissions
About this article
Cite this article
Cheng, Y., Sanderson, C., Jones, M. et al. Low MHC class II diversity in the Tasmanian devil (Sarcophilus harrisii). Immunogenetics 64, 525–533 (2012). https://doi.org/10.1007/s00251-012-0614-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-012-0614-4