
Abstract
Genome and expressed sequence tag information of Xenopus tropicalis suggested that short-consensus repeat (SCR)-containing proteins are encoded by three genes that are mapped within a 300-kb downstream of PFKFB2, which is a marker gene for the regulator of complement activation (RCA) loci in human and chicken. Based on this observation, we cloned the three cDNAs of these proteins using 3′- or 5′-RACE technique. Since their primary structures and locations of the proximity to the PFKFB2 locus, we named them amphibian RCA protein (ARC) 1, 2, and 3. Expression in human HEK293 or CHO cells suggested that ARC1 is a soluble protein of Mr ∼67 kDa, ARC2 is a membrane protein with Mr 44 kDa, and ARC3 a secretary protein with a putative transmembrane region. They were N-glycosylated during maturation. In human and chicken RCA clusters, the order in which genes for soluble, GPI-anchored, and membrane forms of SCR proteins are arranged is from the distant to proximity to the PFKFB2 gene. However, the amphibian ARC1, 2, and 3 resembled one another and did not reflect the same order found in human and chicken RCA genes. This may be due to self-duplication of ARCs to form a family, and it evolved after the amphibia separated from the ancestor of the amniotes, which possessed soluble, GPI-anchored, and membrane forms of SCR protein members. Taken together, frog possesses a RCA locus, but the constitution of the ARC proteins differs from that of the amniotes with a unique self-resemblance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The complement system consists of effectors for foreign cell clearance and regulators for host cell protection (Morgan and Harris 1999). This innate system primarily functions for host defense against foreign pathogens by highlighting target to eliminate (Morgan and Harris 1999). The active fragment C3b of the third component of complement C3 is a main targeting effector conducting complement-mediated host immune response (Morgan and Harris 1999). The effector system is the protease cascade that activates this pivotal membrane-targeting component C3 by cleaving it into C3b and C3a (Morgan and Harris 1999). The active form C3b covalently binds bacterial membrane to alert the presence of invading foreign material to host immune cells (Morgan and Harris 1999). Additional response following C3b deposition is the assembly of membrane attack by hydrophobic molecular association of pore-forming C5b-9 unit (Morgan and Harris 1999). The C3-activating effector scheme is conserved as similar plasma protease system in most of deuterostomes and a part of protostomes (Nonaka and Kimura 2006; Zhu et al. 2005).
Excessive C3 activation often induces consumption of the complement system and damages self tissue, a type of allergy (Morgan and Harris 1999). The C3-step regulatory system has been identified as a family of multifarious proteins with tandemly repeated ∼60 amino acid short consensus repeats (SCRs) (Liszewski et al. 1991). The genes of SCR proteins are clustered at 1q32 called regulator of complement activation (RCA) gene locus in human (Carroll et al. 1988; Liszewski et al. 1991; Rey-Campos et al. 1988). Similar but two split loci of the RCA is found in the mouse (Kingsmore et al. 1989). Furthermore, we found the RCA gene locus with at least four SCR protein genes in the chicken genome (Inoue et al. 2001; Oshiumi et al. 2005). Taken together, the complement regulatory system appears to have developed to diverge into fluid-phase and membrane-bound entities to cope with activation of the complement system. However, phylogenic analysis of the regulatory system is poorly accomplished so far. Our unpublished data show that fish and lamprey possess a single gene encoding a soluble SCR protein in the locus corresponding to the human RCA. Thus, the constituents of the RCA cluster appear different across the vertebrates.
Here, we identified three genes of putative SCR proteins in the Xenopus tropicalis genome. Of these, one is a representative membrane-associated complement regulatory protein. This is the first report on the RCA cluster of amphibia, which may reflect the most ancient form of the cluster of complement regulatory proteins.
Material and methods
Cells and tissues
X. tropicalis was a gift from the National Bio-Resource Project (NBRP) of the MEXT, Japan. Fresh Xenopus organs were isolated from the individual live frogs and then frozen with liquid nitrogen. All samples were stored at –80°C immediately after collection until use. Xenopus blood was collected from the heart by a catheter, and serum was harvested from clotted blood after centrifugation. The human cervical epithelial cell line (HeLa) and Chinese hamster ovary (CHO) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). HEK293FT (human epithelial kidney) cells were obtained from RIKEN Cell Bank (Wako Pure Chemicals, Saitama, Japan). CHO cells were maintained in Ham’s F12/10% fetal calf serum (FCS). HeLa and HEK293FT cells were cultured in MEM/10% FCS and DMEM/10% FCS, respectively. These cells were transfected with cDNAs in expression vectors using the FuGENE HD reagent (Roche) according to manufacture protocol. In some experiments, serum-free medium (Wako Biochemicals, Tokyo, Japan) was used for cell culture, and the supernatants were stored as the source for harvesting transfected gene products in addition to the cell lysates (Kimura et al. 2004). RNaseH was supplied by Promega, Madison WI, USA. ExTaq polymerase was obtained from Takara Bio USA. Marathon cDNA amplification kit was from Clonetech (Palo Alto, CA, USA). FuGENE HD was from Roche Biochemical (Nutely, NJ, USA), and G-Sepahrose was from GE Health care, Madison WI, USA. Block Ace was supplied by Yukijirushi, Sapporo, Japan. Anti-rabbit IGg was obtained from Cappel Laboratories, Cochranville, PA, USA. Neuraminidase and O-glycosidase were from Sigmachemical company, St. Louis, MO, USA and Genayme from Cambridege, MA USA.
Isolation of mRNA and RT-PCR
Total RNA was extracted from Xenopus tissues and cell lines with TRIZOL reagent (Invitrogen) according to manufacture protocol. Four micrograms of total RNA was reverse-transcribed using RNaseH(−) reverse transcriptase and then subjected to 2 min denaturation at 94°C followed by polymerase chain reaction (PCR) cycle of cDNA amplification using ExTaq polymerase for 35 cycle at 94°C 1 min, annealing at 55°C for 1 min followed by 2 min extension at 72°C. The forward and reverse primers used are described in the following section. The products were separated on 1.5 % agarose gels in TAE and identified by ethidium bromide stain.
Cloning of ARC1, 2, and 3
We assembled expressed sequence tag (EST) sequences of ARC1 on the predicted full sequence of ARC1 taken from the DNA database. Primer sequences used for PCR are listed in Table 1. Total RNA extracted from X. tropicalis tissues was used as a template for reverse transcriptase (RT)-PCR for obtaining cDNA. For ARC1, ARC1 primers A, B, C, and D were used (Table 1). We obtained several clones of ARC1 cDNA, and chose a perfect clone without containing PCR errors. During the cloning of the C-terminal region of ARC1 with the primer C and D, we happened to find the short and long cDNA fragments. Aligning the sequences of the two cDNA fragments revealed that the short cDNA sequence lacks the region encoding one SCR domain compared to the long cDNA sequence. We determined the exon/intron structure of ARC1 by comparing the long cDNA sequence with the genome sequence and found that the region absent in the short cDNA fragment of ARC1 exactly corresponds to one exon. Therefore, we concluded that the two cDNA fragments were derived from alternative splicing.
The 5′ region of ARC2 was not found in any EST sequences encoding ARC2; therefore, we carried out 5′ RACE using Marathon cDNA Amplification kit with AP1, AP2, ARC2 A′, and ARC2 A primers (Table 1). Based on the result of sequencing, we finally cloned a cDNA-encoding full length ARC2 using ARC2 B and C primers.
The 5′ and 3′ end of ARC3 ORF was not found in any ARC3 EST sequences. To determine the full length ARC3 sequence, we executed 5′ and 3′ RACE using AP1, AP2, ARC3 A, ARC3 A′, ARC3 B, and ARC3 B′ as primers (Table 1). Based on the obtained ARC3 sequence, we cloned the cDNA encoding the full length ARC3 ORF with ARC3 C, E, D, and F primers.
The conditions of nested PCR were described in a previous report (Inoue et al. 2001; Oshiumi et al. 2005). These cDNA clones were ligated into the Xho/NotI site of pEFBOS expression vector with HA-tag at the C-terminal ends.
Immunoprecipitation, SDS-PAGE, and Western blotting
Immunoprecipitation was performed using the supernatants of HEK293FT cells transfected with plasmids by FuGENE HD. After incubation for 24 h at 37°C, 2 ml of the supernatant was incubated with 50 μl of Protein G-Sepharose for 1 h at 4°C to remove nonspecific proteins. The cleared supernatants were mixed with 0.5 μg of rabbit anti-HA antibody (Ab) and 20 μl of Protein G-Sepharose beads. The mixture was incubated for 12 h at 4°C. The beads were washed thrice in the wash buffer [phosphate-buffered saline (PBS)/0.02% NP-40] and the beads were extracted with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. The samples were subjected to SDS-PAGE followed by Western blotting as described previously (Kimura et al. 2004).
We detected the secreted ARC proteins from the cell culture supernatant. The HEK293 cells were transfected in six-well plate with the plasmid encoding ARC proteins, using FuGENE HD. After 24 h, 2 ml of the culture medium was collected. To remove proteins that nonspecifically bound the Sepharose beads, we added Proteins G Sepharose (50 μl, prewashed) to the medium, and then the protein G Sepharose-containing medium was rotated at 4°C for 1 h. The medium was centrifuged at 2,000 rpm for 1 min, and the supernatant was moved to a new tube. Anti-HA rabbit polyclonal antibody and prewashed protein G Sepharose were added to the tube, and the tube was rotated at 4°C for 24 h. The protein G Sepharose was collected by centrifugation and then washed three times with wash buffer. The immunoprecipitated samples were extracted with SDS-PAGE sample buffer by boiling for 5 min. The samples was analyzed by SDS-PAGE and visualized by Western blotting. We could detect secreted ARC1 and ARC3 proteins.
Immunofluorescence analysis of transfected cells
HeLa cells expressing HA-tag-labeled ARC proteins were incubated with 100 µl of 2 µg/ml rabbit anti-HA Ab for 1 h at 37°C in PBS containing 1% (w/v) bovine serum albumin. The cells were washed, incubated with a 1:100 dilution of Alexa-conjugated anti-rabbit IgG Ab for 30 min at 37°C in PBS containing 10% (w/v) Block Ace, washed, and mounted on glass slides in PBS containing 2.3% 1,4-diazabiccyclo-2-octane and 50% glycerol. The stained cells were visualized at ×40 magnification under a FLUOVIEW (Olympus, Tokyo, Japan). Images were captured using the attached computer software, FLUOVIEW.
Deglycosylation analysis
The methods for analyses using deglycosidases were described previously (Kimura et al. 2004). Briefly, transfectants (5 × 106) were solubilized in 50 mM Tris-maleate (pH 8.6) containing 1% Nonidet P-40, 10 mM EDTA, 1 mg/ml iodoacetamide, 1 mM phenylmethylsulfonyl fluoride (PMSF) for O-glycosidase analysis. For N-glycosidase analysis, the same buffer except 20 mM Tris-maleate (pH 6.0) was used. Solubilized proteins were centrifuged at 15,000 rpm for 30 min at 4°C, the pellets were removed, and the supernatants were incubated with 100 µU of neuraminidase for 1 h at 37°C. Then, the samples were treated with either 250 mU of N-glycosidase or 1 mU of O-glycosidase for 16 h at 37°C. The samples were subjected to SDS-PAGE followed by immunoblotting. ARC proteins were detected with anti-HA Ab as described above.
Protein domain structure and homology analyses
The domain structures of Xenopus proteins were predicted using SMART program (http://smart.embl-heidelberg.de/). Signal peptide was predicted by SignalP program (http://www.cbs.dtu.dk/services/SignalP/) (Emanuelsson et al. 2007) using the hidden Markov or neural network model. Although the hidden Markov model failed to predict the ARC3 signal peptide, the neural network model predicted it. Homologies between Xenopus and chicken or human proteins were examined by BLAST search analysis. Homologies among SCR domains were determined by comparing the SCR domains of chicken proteins with those of human proteins using TBLASTN program in NCBI BLAST server and GENETYX-MAC Ver. 11.2.1 (GENETYX) maximum matching program. The N-glycosylation sites were predicted using NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/).
Results
RCA locus in X. tropicalis
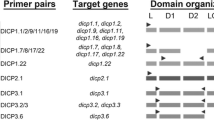
Genes in the RCA locus are closely linked to those of PFKFB2 in human, mouse, and chicken (Oshiumi et al. 2005). We searched for the RCA locus in the X. tropicalis genome (JGI genome server) by in silico analysis using the human PFKFB2 full-length sequence as the probe. A X. tropicalis gene sequence similar to that of human PFKFB2, but not other family members, was found by TBLASTN search against human genome database. Furthermore, three genes containing putative SCRs were identified in close proximity 3′ to the PFKFB2 gene (Fig. 1a,c). A majority of the SCR-coding regions in these genes were encoded by single exons. The predicted amino acid sequences of all the three genes contain typical SCRs, similar to human and chicken complement regulatory proteins (Fig. 1b). These properties support the existence of a RCA cluster of complement regulatory proteins in frog in a fashion similar to that of human and chicken (Fig. 1c). We call the RCA of amphibia genes ARC and named them from the proximity to the PFKFB2 locus, ARC1, ARC2, and ARC3.



Identification of three Xenopus SCR proteins. a Structures of the X. tropicalis ARC genes in the genome. The prediction of exon and intron was followed to the JGI database. The ag-gt consensus sequences for splicing are conserved. Non-cording regions and cording regions are represented as open and grayed rectangles, respectively. Putative transmembrane (TM) portions are shown as closed rectangles. b Each SCR sequence of ARC1, 2, and 3 was compared with that of human or chicken SCR proteins using GENETYX ver 11. 2.1 maximum matching program. Regions with high homology are shown as red (>45%), orange (45∼40%), brown (40∼35%), or yellow (30∼35%). c Comparison of the frog RCA locus with the human and chicken RCA loci. According to the X. tropicalis genome sequence, ARC1, 2, and 3 are clustered in the ∼300-kbp region, which is longer than the G. gallus RCA but shorter than the human RCA locus. The RCA loci of these three species are linked with the PFKFB2 gene, and the gene directions are also conserved. In both human and chicken RCAs, soluble regulators, C4 bp and CRES, are most proximal to PFKFB2, and membrane proteins, MCP and CREM, are most distal to PFKFB2. In Xenopus, the soluble protein, ARC1, is most proximal to PFKFB2 gene as human C4 bp and chicken CRES, but unlike human and chicken RCAs, we could not find any GPI-anchored protein in the Xenopus RCA locus
We have determined the gene structures including the exon–intron boundaries of these three frog RCA genes (Figs. 1a and 2). The results show that SCR2 of ARC1 and ARC2, and SCR2, SCR3, SCR7, and SCR8 of ARC3 were encoded by split exons (Fig. 1a). The splitting features of SCR2 of ARC 1 and 2 and SCR3 of ARC3 were similar to the functionally essential exons of the human and chicken complement regulatory proteins. Furthermore, amino acid sequences of split exon-encoded SCR2 or SCR3 of ARC1, 2, and 3 were highly similar (up to 40%) to those of corresponding functional SCRs of chicken CRES (Fig. 1a,b). Further, the divisions in their coding regions occur at similar positions. However, other chicken RCA proteins CREG and CREM did not show such similarity with ARC proteins (Oshiumi et al. 2005). Thus, it is likely that the split exons in the SCR2 of ARC1/2 and SCR3 of ARC3 encode functionally active domains.



Complete amino acid sequences of ARC1, 2, and 3. Deduced amino acid sequences of ARC1 (a), ARC2 (b), and ARC3 (c) are shown under the nucleotide sequences. Asterisks indicate the stop codons. The predicted signal sequences are underlined. The amino acid sequences with hydrophobicity (putative transmembrane portions) are double underlined in ARC2 and 3. The broken lines show the position of exon/intron boundaries. The rectangles show the predicted N-linked glycosylation Asn residues. The nucleotide sequences have been registered in the EMBL Data Library/GenBank/DDBJ databases with the accession numbers AB474590 (ARC1), AB474591 (ARC2), and AB474592 (ARC3)
Molecular cloning of ARC1, 2, and 3
EST sequences published covered the whole ORF of ARC1 and a 3′ part of ARC2. ARC3 EST was only partially identified. In order to obtain the complete ORF of ARC2, nested PCR with 5′ RACE was performed. We obtained products whose sequences matched the EST sequences containing an upstream ATG translation initiation and stop codons. To clone the complete ARC3 cDNA, we performed 5′ and 3′ RACEs and obtained the ARC3 cDNA sequence containing the 5′ ATG start and the 3′ stop codons. The sequences of ARC1, 2, and 3 are shown in Fig. 2a, b, and c (AB474590, AB474591, AB474592). SignalP analysis suggested that ARC1, ARC2, and perhaps ARC3 have signal sequences. ARC2 and 3 have putative transmembrane regions of ∼20 amino acids. The properties of the predicted ARC proteins together with the results of PSORT analysis suggested that ARC1 is a secretary or cytoplasmic protein, while ARC2 and 3 are type I membrane proteins.
From the sequence, ARC1 (later named ARC1L) was found to be 470 amino acid soluble protein with seven SCRs. ARC2 was a 329 amino acid membrane protein with four SCRs, and ARC3 was a 563 amino acid membrane protein with eight SCRs.
Tissue distribution and cellular localization of ARCs
Tissue distribution profiles of these ARC messages were examined by RT-PCR. RNA samples were extracted from the organs indicated and the cDNAs used for templates. Amplifiable sequences of ARC1 (518–1,447 bp), ARC2 (1–987 bp), and ARC3 (1–644 bp) were selected for RT-PCR analysis. We designed PCR primers based on the derived sequences (Table 1). The results showed that the mRNAs of ARC1 and 3 were ubiquitously expressed, while the ARC2 mRNA was detected only in the liver, intestine, and muscle (Fig 3a). We found a faster mobility band below the predicted ARC1 band in all lanes, suggesting the presence of a splicing variant in ARC1. The short splicing variant was predicted to encode a protein with six SCRs, which we named ARC1s, as described in the “Materials and methods” section.

Expression analyses of the ARC mRNAs in adult frog tissues. a RT-PCR analysis. The ARC cDNAs were amplified using the same PCR procedure (see the “Materials and methods” section). Actin was used for a positive control. No product was amplified with the actin cDNA from nonreverse transcribed samples. The experiments were performed by two independent samples, and the representative data is shown. b Imaging analysis of ARCs in human cells. HeLa cells expressing HA-tag-labeled ARC proteins were stained with anti-HA Ab and Alexa-conjugated goat anti-rabbit IgG Ab. Cells were fixed and observed with confocal microscope
We subcloned HA-tagged ARC1L, 2, and 3 using the cloned ARC sequences as templates. When the plasmids were transfected into HEK293FT and CHO cells, the ARC proteins were detected in cell lysates by Western blotting using anti-HA Ab. By confocal analysis using anti-HA Ab, ARC1L and ARC3 were localized to the cytoplasm (Fig. 3b). ARC2 was localized to the cell-surface membrane, supporting the prediction from its amino acid sequence to be a type I membrane protein (Fig. 3b). We found that the transfected cells secreted ARC1 and 3 proteins into the supernatants. Since the ARC3 protein showed higher molecular weight in the supernatant (75 kDa) than in the cell lysate (70 kDa), it is not yet certain if ARC3 is proteolytically cleaved out from the membrane. ARC3 may retain as an unprocessed form in the cytoplasmic granules and gradually mature during secretion into the media irrespective of the presence of the hydrophobic transmembrane-like sequence.
Posttranslational sugar modification in ARCs
Human complement regulatory proteins often undergo posttranslational sugar modification. Since the molecular masses of the expressed ARC proteins estimated by SDS-PAGE were higher than those predicted from the primary structures, we tested their sugar moieties. N-glycosidase treatment of ARC1L (67 kDa), ARC2 (44 kDa), and ARC3 (75 kDa) reduced the molecular masses to 55, 40, and 72 kDa, respectively (Fig. 4). The two band patterns observed in N-glycosidase-treated ARC2 and ARC3 may reflect either incomplete sugar digestion or heterogeneous sugar compositions. N-linked sugar modifications of ARC1, 2, and 3 during maturation are also supported by NetNGlyc 1.0 (Julenius et al. 2005; Fig. 2). NetOglyc 3.1 (Blom et al. 2004) analysis did not support the presence of O-linked sugar in ARC proteins. Thus, ARC proteins undergo N-linked sugar modification, which appears to be a process linked to their maturation. On SDS-PAGE, the mature forms of ARC1L (lane 5 of Fig. 4) and ARC3 (lane 13 of Fig. 4) exhibited single bands, but ARC2 showed multiple bands (lane 9 of Fig. 4) reflecting possible multiple modifications for this protein. Interestingly, ARC1L and ARC3 secretary forms have almost identical molecular sizes to their cytoplasmic forms. The mechanism of secretion of ARC3 from transfected cells is yet to be determined.

Deglycosylation analysis of ARCs. Immunoblotting profiles of ARC1, 2, and 3. Cell lysates containing ARC proteins were treated with N- or O-glycosidase and analyzed on SDS-PAGE and immunoblotting. Arrows indicate major bands of ARC1L, ARC2, and ARC3. lanes 1–4 control with no sample, lanes 5-8 ARC1L, lanes 9–12 ARC2; lanes 13–16 ARC3
Phylogenetic analysis of ARCs
Previously, we showed that avian, Gallus gallus, RCA genes, CRES, CREM, and CREG, were homologs of C4 bp, MCP, and DAF, respectively (Oshiumi et al. 2005), despite their frequent domain shuffling among those genes. To examine the orthologous relationship between human and amphibian RCA genes, their protein sequences were aligned by ClustalW software, and the phylogenic tree was drawn by neighbor-joining method. Surprisingly, we found that the amphibian ARC1, 2, and 3 are closely related with each other and not clustered to contain any ortholog of the human RCA gene (Fig. 5a). To further confirm that the amphibian RCA proteins are more similar to each other than to human RCA proteins, we carried out BLAST search analyses. The results showed that ARC1 was more similar to ARC2 and 3 than human RCA genes. ARC2 and 3 also resemble more to ARC1 than human RCA (Table 2). These results support the notion that X. tropicalis RCA genes underwent duplication after X. tropicalis ancestor had diverged from human and their common ancestor.

Phylogenic analysis of frog ARCs in comparison with chicken, mouse and human complement regulatory proteins. a The protein sequences of the complement regulatory proteins were aligned with the ClustalW program, and the phylogenic tree was made by neighbor-joining method using the DDBJ server (http://clustalw.ddbj.nig.ac.jp/top-j.html). Number on each node represents bootstrap probability that is 1,000 times reiteration. Lacrep lamprey complement regulatory protein (AB061219), Crry mouse complement regulatory protein (NP_038527), CRES (BAE16761), CREG (BAE16762), CREM (BAB16878) are complement regulatory proteins of chicken. b Each SCR of ARCs amino acids sequences were compared using the ClustalW program, and the phylogenic tree was drawn by neighbor-joining method. Four clades were obtained with this analysis and named clade A, B, C, and D. c The amino acids sequence identity between the SCRs of ARCs. According to the results in B, each SCR domain of ARC1 is annotated as A-B-C-D-A-C-C from the N-terminal end. ARC2 SCR domains are annotated as A-B-C-D. The C-terminal two SCR domains of ARC3 could not be specified into one clade, but the two SCR domains show similarity to the SCR domains of B and C clades, and thus, we classified the two as a clade of BC. From the N-terminal end, the SCR domains of ARC3 were annotated as A-A-B-C-D-A-BC-BC. The amino acids identities of SCR domains of the same clade, which are classified in B, were calculated
The most popular form of the human CR1 protein consists of four tandem repeats of a unit of seven SCR domains (Klickstein et al. 1987). To elucidate whether X. tropicalis possesses an SCR repeat-containing gene in the RCA locus similar to that of human CR1, the sequences of each SCR domain of ARCs were compared to each other (Fig. 5b and Table 2). The results showed that almost all SCR domains of X. tropicalis RCA proteins could be clustered into four groups. We named these four SCR domains SCR-A, B, C, and D. The order of ARC1 SCR is A-B-C-D-A-C-C (Fig. 5c). In ARC2 protein, there is no duplicate of SCR domain, and its order is A-B-C-D (Fig. 5c). In ARC3, two ambiguous SCR domains follow A-A-B-C-D of SCR domains (Fig. 5c). Because the order, SCR-A-B-C-D, commonly exists in the three ARC proteins, the ancestral ARC protein seems to consist of SCR-A-B-C-D, and the duplication of SCR domains might have occurred in ARC1 and ARC3, but not ARC2. Next, we compared the similarity of ARC SCR-A, B, C, D to human or chicken RCA protein SCRs. Interestingly, the ARC SCRs were similar to DAF or CREG proteins, both of which are GPI-anchored proteins, in that DAF and CREG fundamentally consist of SCR-A-B-C-D. This is reminiscent of the order of SCR-A-B-C-D found in the putative Xenopus ARC ancestor (Fig. 1b).
Discussion
Here we demonstrated that X. tropicalis possesses three SCR proteins ARC1, 2, and 3. They were mapped downstream of the PFKFB2 gene, like the RCA loci of human and chicken. In human, group B complement regulatory proteins, C4 bp, DAF, CR2, CR1, and MCP (Krushkal et al. 2000), clustered downstream of PFKFB2 in chromosome 1q32 (Rey-Campos et al. 1988). In chicken, CRES, CREG, CR1-like undefined gene, and CREM are clustered downstream of PFKFB2 in a microchromosome (Oshiumi et al. 2005). Thus, the order of soluble, GPI-anchored, and membrane forms of RCA genes is essentially the same in human and chicken. We expected that ARC1, 2, and 3 reflect the order of CRES, CREG, and CREM in chicken. However, ClustalW alignment and expression analyses showed that amphibian ARCs did not follow the conventional organization. The three ARC proteins resemble each other. From these current views, we speculate that ARCs self-duplicated to form a RCA family. The ARC family evolved after the amphibia separated from the ancestor of the homeotherm, which possess soluble, GPI-anchored, and membrane forms of SCR protein members.
Human and chicken RCA products have complement regulatory activity (Morgan and Harris 1999; Oshiumi et al. 2005) by either accelerating the decay of the C3 convertases or cleaving C3b into inactive forms. It is not surprising that the frog ARC proteins possess complement regulatory activity toward the frog C3b and C3 convertases, the presence of which has been reported (Fujii et al. 1985; Grossberger et al. 1989; Sekizawa et al. 1984). Although the functional point of ARCs needs to be experimentally addressed, gene structure analysis suggested that SCR2 of ARC1 (s and L forms), ARC2, and ARC3 were encoded by split exons similar to those of human/chicken C3-step regulatory proteins. In human RCA proteins, SCR2 play a central role in C3b inactivation (Casasnovas et al. 1999; Liszewski et al. 2000). Hence, the primary composition of SCR2 is conserved across human and frogs.
This study further revealed that frog has a membrane-associated form of RCA proteins. ARC2 and ARC3 possessed transmembrane domains. By overexpression analysis, ARC2 protein was localized to the surface of cell membrane, which confirms the notion that ARC2 is a membrane protein. Many reports suggested that mammals and chicken do possess membrane forms of complement regulatory protein (Morgan and Harris 1999; Oshiumi et al. 2005). However, no report indicated that fish have a membrane SCR protein, although we showed that they have soluble SCR proteins for inhibiting fluid-phase complement activation (Oshiumi and Seya, unpublished data). In fact, teleost fish have single or duplicated genes encoding soluble SCR proteins around the downstream locus of PFKFB2. We favor a tentative propose that amphibia are the first vertebrates to possess membrane-associated RCA proteins.
From its structural analysis, we are prone to think that ARC3 could also be a membrane-associated SCR protein which gets clipped by proteolytic cleavage to generate a secreted form. How the ARC3 soluble form naturally generates in human cells remains undetermined. The discrepancy observed in ARC3 cellular vs. soluble proteins needs further analyses.
Since ARC1 and 3 were ubiquitous while ARC2 had limited expression in the liver and intestine, functional divergence might have occurred in amphibian RCA proteins. There are several tyrosine-based motifs in the cytoplasmic tail of ARC2. As its functionality may be mediated by the tail sequences, ARC2, unlike the others, may have a special role in complement-mediated immune response. In humans, CD46 (MCP) and CD55 (DAF) are ubiquitously expressed membrane proteins that protect host cells from complement-mediated cell damage (Atkinson 1996), while CD35 (CR1) and CD21 (CR2) confer cell type-specific expression and specified functions (Ahearn and Fearon 1989). CD46 may be a signal-transducing receptor, presumably via the cytoplasmic tyrosines (Crimeen-Irwin et al. 2003; Kemper et al. 2005).
Lack of the ubiquitously expressed membrane protein in the frog RCA may predict the presence of additional host-protective proteins in other loci of frog. Search by TBLASTN in itself did not enable us to identify genes with SCRs as we could previously succeed with the chicken genome (Oshiumi et al. 2005). The presence of many introns within the genes and low numbers of consensus amino acids in each SCR complicate the search. Therefore, further analyses besides BLASTN searches are required for discovery of additional SCR genes.
We cloned two messages that encode the isoforms of ARC1 protein, ARC1s and ARC1L by chance. The SCR6 and 7 in ARC1 are >90% homologous to each other, and the primers we selected permitted the cloning of these two forms. Exon duplication may have occurred lately in the ARC1 gene to yield ARC1s and ARC1L via alternative splicing. Thus, continuous gene duplication in the RCA locus occurs that facilitates the generation of various lengths of SCR proteins. In fact, there are variable-sized CR1 proteins in humans (Dykman et al. 1983; Dykman et al. 1984). Further search for additional SCR proteins in frog will be an important aspect of future research in this area.
The present study adds more information to the existing knowledge of the origin of RCA gene cluster. Our scheme suggests that the prototype of the RCA locus contains SCR domains A, B, C, and D. ARC1, 2, and 3 are independently generated through gene duplication after the ancestor of amphibia separated from that of mammals. The mammalian and chicken RCA clusters essentially contain membrane type, GPI-anchored type, and soluble type of SCR protein genes in the same order (Fig. 1c). This fundamental architecture of RCA appears to be established through gene duplication and independent evolution. Thus, the repertoire formation in the RCA locus of frog and other higher vertebrates is modally different. We prefer to interpret that the frog RCA locus originated from a single RCA gene present in fish or jawless fish (Kimura et al. 2004), which probably is the cause for the three ARC genes to have close resemblance to one another. In either case, the split exons for SCR2 are highly conserved, suggesting that this split exon motif is rooted in the prototype of the ancestor RCA protein.
We want to focus on the gap in the RCA properties between amniotes and aqueous vertebrates. Microbial environment in water is different from that in land (Matsuo et al. 2008; Seya et al. 2009). The Toll-like receptor (TLRs) system has been developed to protect fish against Gram-negative bacteria and double-stranded RNA viruses, which live in water environment (Matsuo et al. 2008; Oshiumi et al. 2003; Seya et al. 2009). In general, complement exerts strong cytolytic activity toward Gram-negative bacteria and enveloped viruses. Thus, the water microbes may activate the complement system, too. We therefore infer that natural selection happened to maintain species-specific RCA cluster. This may be a reason why some water invertebrates retain the complement system (Zhu et al. 2005). On land, Gram-positive bacterial and mono-nega RNA virus infections are also prevalent in many vertebrates. In these cases, complement regulatory proteins are indispensable for coping with robust complement activation secondary to infection. The complement gene disruptions invariably lead to severe autoimmune aberrance secondary to infection in mammals (Morgan and Harris 1999). Broad repertoire of complement regulatory proteins is needed for land life to circumvent irregular complement-related disorders (Ahearn and Fearon 1989; Atkinson 1996; Morgan and Harris 1999). Since the complement system is not merely a host defense against microbes, more information about other innate systems (Oshiumi et al. 2009; Seya et al. 2009) is required to assess the importance of evolution of complement and complement regulatory proteins for terrestrial life.
References
Ahearn JM, Fearon DT (1989) Structure and function of the complement receptors, CR1 (CD35) and CR2 (CD21). Adv Immunol 46:183–219. doi:10.1016/S0065-2776(08) 60654-9
Atkinson JP (1996) Impact of the discovery of membrane inhibitors of complement. Res Immunol 147:95–100. doi:10.1016/0923-2494(96) 87180-X
Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649. doi:10.1002/pmic.200300771
Carroll MC, Alicot EM, Katzman PJ, Klickstein LB, Smith JA, Fearon DT (1988) Organization of the genes encoding complement receptors type 1 and 2, decay-accelerating factor, and C4-binding protein in the RCA locus on human chromosome 1. J Exp Med 167:1271–1280. doi:10.1084/jem.167.4.1271
Casasnovas JM, Larvie M, Stehle T (1999) Crystal structure of two CD46 domains reveals an extended measles virus-binding surface. EMBO J 18:2911–2922. doi:10.1093/emboj/18.11.2911
Crimeen-Irwin B, Ellis S, Christiansen D, Ludford-Menting MJ, Milland J, Lanteri M, Loveland BE, Gerlier D, Russell SM (2003) Ligand binding determines whether CD46 is internalized by clathrin-coated pits or macropinocytosis. J Biol Chem 278:46927–46937. doi:10.1074/jbc.M308261200
Dykman TR, Cole JL, Iida K, Atkinson JP (1983) Structural heterogeneity of the C3b/C4b receptor (Cr 1) on human peripheral blood cells. J Exp Med 157:2160–2165. doi:10.1084/jem.157.6.2160
Dykman TR, Hatch JA, Atkinson JP (1984) Polymorphism of the human C3b/C4b receptor. Identification of a third allele and analysis of receptor phenotypes in families and patients with systemic lupus erythematosus. J Exp Med 159:691–703. doi:10.1084/jem.159.3.691
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protocols 2:953–971. doi:10.1038/nprot.2007.131
Fujii T, Sekizawa A, Katagiri C (1985) Characterization of the fourth component of complement in the serum of the clawed frog Xenopus laevis. Immunology 56:743–750
Grossberger D, Marcuz A, Du Pasquier L, Lambris JD (1989) Conservation of structural and functional domains in complement component C3 of Xenopus and mammals. Proc Natl Acad Sci USA 86:1323–1327. doi:10.1073/pnas.86.4.1323
Inoue N, Fukui A, Nomura M, Matsumoto M, Nishizawa Y, Toyoshima K, Seya T (2001) A novel chicken membrane-associated complement regulatory protein: molecular cloning and functional characterization. J Immunol 166:424–431
Julenius K, Molgaard A, Gupta R, Brunak S (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15:153–164. doi:10.1093/glycob/cwh151
Kemper C, Verbsky JW, Price JD, Atkinson JP (2005) T-cell stimulation and regulation: with complements from CD46. Immunol Res 32:31–43. doi:10.1385/IR:32:1-3:031
Kimura Y, Inoue N, Fukui A, Oshiumi H, Matsumoto M, Nonaka M, Kuratani S, Fujita T, Seya T (2004) A short consensus repeat-containing complement regulatory protein of lamprey that participates in cleavage of lamprey complement 3. J Immunol 173:1118–1128
Kingsmore SF, Vik DP, Kurtz CB, Leroy P, Tack BF, Weis JH, Seldin MF (1989) Genetic organization of complement receptor-related genes in the mouse. J Exp Med 169:1479–1484. doi:10.1084/jem.169.4.1479
Klickstein LB, Wong WW, Smith JA, Weis JH, Wilson JG, Fearon DT (1987) Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J Exp Med 165:1095–1112. doi:10.1084/jem.165.4.1095
Krushkal J, Bat O, Gigli I (2000) Evolutionary relationships among proteins encoded by the regulator of complement activation gene cluster. Mol Biol Evol 17:1718–1730
Liszewski MK, Post TW, Atkinson JP (1991) Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu Rev Immunol 9:431–455. doi:10.1146/annurev.iy.09.040191.002243
Liszewski MK, Leung M, Cui W, Subramanian VB, Parkinson J, Barlow PN, Manchester M, Atkinson JP (2000) Dissecting sites important for complement regulatory activity in membrane cofactor protein (MCP; CD46). J Biol Chem 275:37692–37701. doi:10.1074/jbc.M004650200
Matsuo A, Oshiumi H, Tsujita T, Mitani H, Kasai H, Yoshimizu M, Matsumoto M, Seya T (2008) Teleost TLR22 recognizes RNA duplex to induce IFN and protect cells from birnaviruses. J Immunol 181:3474–3485
Morgan BP, Harris CL (1999) Complement regulatory proteins. Academic Press, 1-382
Nonaka M, Kimura A (2006) Genomic view of the evolution of the complement system. Immunogenetics 58:701–713. doi:10.1007/s00251-006-0142-1
Oshiumi H, Tsujita T, Shida K, Matsumoto M, Ikeo K, Seya T (2003) Prediction of the prototype of the human Toll-like receptor gene family from the pufferfish, Fugu rubripes, genome. Immunogenetics 54:791–800
Oshiumi H, Shida K, Goitsuka R, Kimura Y, Katoh J, Ohba S, Tamaki Y, Hattori T, Yamada N, Inoue N, Matsumoto M, Mizuno S, Seya T (2005) Regulator of complement activation (RCA) locus in chicken: identification of chicken RCA gene cluster and functional RCA proteins. J Immunol 175:1724–1734
Oshiumi H, Matsumoto M, Seya T (2009) Molecular evolution of complement regulatory proteins. Comp Funct Genomics (in press)
Rey-Campos J, Rubinstein P, Rodriguez de Cordoba S (1988) A physical map of the human regulator of complement activation gene cluster linking the complement genes CR1, CR2, DAF, and C4BP. J Exp Med 167:664–669. doi:10.1084/jem.167.2.664
Sekizawa A, Fujii T, Katagiri C (1984) Isolation and characterization of the third component of complement in the serum of the clawed frog, Xenopus laevis. J Immunol 133:1436–1443
Seya T, Matsumoto M, Ebihara T, Oshiumi H (2009) Functional evolution of the TICAM-1 pathway for extrinsic RNA sensing. Immunol Rev 227:44–53. doi:10.1111/j.1600-065X.2008.00723.x
Zhu Y, Thangamani S, Ho B, Ding JL (2005) The ancient origin of the complement system. EMBO J 24:382–394. doi:10.1038/sj.emboj.7600533
Acknowledgments
This work was supported in part by CREST and Innovation, Japan Science and Technology Corporation (JST), the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, MEXT, Sapporo Biocluster “Bio-S”, the Knowledge Cluster Initiative of the MEXT, Grants-in-Aid from the Ministry of Education, Science, and Culture (Specified Project for Advanced Research) and the Ministry of Health, Labor, and Welfare of Japan, Mitsubishi Foundation, Mochida Foundation, NorthTec Foundation, Yakult Foundation, and Takeda Foundation. Providing us with X. tropicalis from the National Bio-Resource Project (NBRP) of the MEXT is gratefully acknowledged. Thanks are also due to Drs. A. Ishii, A. Matsuo, and other laboratory members for critical discussions, and to Dr. V. Kumar (St. Louis Univ.) for his English edition.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oshiumi, H., Suzuki, Y., Matsumoto, M. et al. Regulator of complement activation (RCA) gene cluster in Xenopus tropicalis . Immunogenetics 61, 371–384 (2009). https://doi.org/10.1007/s00251-009-0368-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-009-0368-9