
Abstract
We have identified the gene coding for a novel serine protease with close similarities to mammalian granzymes from nonspecific cytotoxic cells of a teleost fish Oreochromis niloticus. The genomic organization of tilapia granzyme-1 (TLGR-1) has the signature five-exon–four-intron structure shared by all granzymes and similar hematopoietic Ser proteases. Molecular modeling studies suggested a granzyme-like structure for this protein with four disulfide linkages and two additional Cys residues. The expression of this gene is found to be restricted to cytotoxic cell populations with a low level of constitutive expression when compared to similar granzymes in other teleost species. High levels of transcriptional activation of TLGR-1 with different stimuli suggested that this gene is highly induced during immune reactions. Triplet residues around the active site Ser of TLGR, which determines the primary substrate specificity of granzymes, differ significantly from that of other granzymes. Recombinant TLGR-1 was expressed in the mature and proenzyme forms using pPICZ-alpha vector in the Pichia pastoris expression system. Recombinant TLGR-1 was used to determine the primary substrate specificity of this protease using various synthetic thiobenzyl ester substrates. In vitro enzyme kinetics assays suggested a preference for residues with bulky side chains at the P1 site, indicating a chymase-like activity for this protease. These results indicate the presence of novel granzymes in cytotoxic cells from ectothermic vertebrates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Granzymes belong to a family of serine proteases, which constitute the major components of granules of professional killer cells such as cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells. Along with perforin, a pore-forming protein that is also found in these granules, granzymes play a major role in inducing target cell death (Lieberman 2003; Russell and Ley 2002; Trapani 2001). All the mammalian granzymes described so far are highly conserved and are located in three clusters on three chromosomes (Grossman et al. 2003). Granzymes A and B are the most extensively studied granzymes. The different pathways used by granzymes A and B to initiate cytotoxicity have been described (Lieberman and Fan 2003; Trapani and Sutton 2003). However, novel mechanisms of cytotoxicity are being discovered for these granzymes, expanding our knowledge about complex pathways involved in cell death and proliferation (Beresford et al. 2001; Fan et al. 2002, 2003a,b; Han et al. 2004; Sebbagh et al. 2005). The function of many of the other granzymes is not well understood, thus they are collectively called as orphan granzymes (Grossman et al. 2003).
Nonredundant pathways of cell-mediated cytotoxicity have been proposed for granzymes, suggesting a ‘fail-safe’ mechanism provided by multiple granzymes (Grossman et al. 2003; Lieberman 2003). The role of granzymes in inducing target cell death has been investigated extensively in vitro (Trapani and Smyth 2002). However, advances in in vivo studies demonstrating the significance of individual granzymes in cytotoxicity lag behind. Targeted gene disruption studies of granzymes have resulted in focal immune deficiencies (Ebnet et al. 1995; Heusel et al. 1994; Trapani and Smyth 2002; Wilharm et al. 1999b). Recent attempts to understand the individual effects of members of the granzyme B gene cluster has revealed the significant role played by orphan granzymes present downstream of the granzyme B gene in mice (Revell et al. 2005). Such results indicate the relative significance of other granzymes that have not been fully investigated.
Granzymes are classified based on their primary substrate specificities, and four enzymatic activities have been described: tryptase, Asp-ase, Met-ase, and chymase. The most extensively studied mammalian granzymes fall under three of the above specificities. Granzymes A and K have tryptase activity, granzyme B has Asp-ase activity, while granzyme M has Met-ase activity (Kam et al. 2000). Many of the orphan granzymes have chymase activity.
The presence of multiple granzymes can be interpreted as a result of molecular strategies developed by the immune system to compete with pathogens. These pathogens have coexisted and coevolved with immune systems of organisms for millions of years (Trapani and Smyth 2002). Genetic comparisons and chromosomal clustering of granzymes suggest gene duplication events as a means of generating newer granzymes (Grossman et al. 2003). Almost all the assumptions on granzyme evolution are based on the gene sequence information from human and rodents due to the lack of sequence information in other organisms. The availability of nonmammalian granzyme sequences would add new insights into the evolution of these proteases with very narrow substrate specificity, and it would also aid in the discovery of newer killing pathways (Hink-Schauer et al. 2003; Praveen et al. 2004).
Nonspecific cytotoxic cells (NCC) are the first identified cytotoxic cell population in teleosts. First described in the channel catfish (Graves et al. 1985), these cells have been well characterized in a number of lower vertebrates (Jaso-Friedmann et al. 2002; Jaso-Friedmann and Evans 1999; McKinney and Schmale 1994; Suzumura et al. 1994). Although first described as agranular (Graves et al. 1985), NCC in trout, tilapia, and catfish do have small granules, and contact with target cells leads to granule exocytosis with necrotic and apoptotic death pathways (Faisal et al. 1989; Greenlee et al. 1991; Jaso-Friedmann et al. 1990). The requirement for calcium for the induction of cytotoxicity by NCC was an early indication that these cells use the granule exocytosis killing pathway (Carlson et al. 1985). We have previously reported the existence of granzyme-like serine proteases in the NCC of channel catfish (Ictalurus punctatus) and tilapia (Oreochromis niloticus) (Praveen et al. 2004). Here, evidence is presented for the full-length sequence of the cDNA and gene for a granzyme-like protease from tilapia, which was designated as tilapia granzyme-1 (TLGR-1), its expression pattern, and the substrate specificity of the recombinant protein. These results demonstrate the molecular characterization of a granzyme from a cytotoxic cell of nonmammalian vertebrates.
Materials and methods
Cloning of full-length TLGR-1
The identification and partial sequence of a granzyme-like serine protease from a tilapia NCC cDNA library have been previously reported (Praveen et al. 2004). Full-length sequence of that tilapia granzyme (TLGR-1) was obtained by rapid amplification of cDNA ends (RACE). Fresh RNA was purified from tilapia NCC from peripheral blood for the RACE as described before (Praveen et al. 2004). Briefly, mRNA (500 ng) was reverse-transcribed with granzyme-specific primers using Generacer Superscript II RT module (Invitrogen, CA). First strand cDNA was subjected to homopolymeric tailing with cytosine using terminal transferase (Roche). Later, dC-tailed cDNA was amplified using abridged anchor primers (Invitrogen), and gene-specific nested primers followed by a reamplification using a nested abridged universal amplification primer and another nested gene-specific primer (Praveen et al. 2004). The amplicons were purified and TA-cloned for sequencing. For 3′ RACE, RNA was reverse-transcribed using an anchor primer, EPB-18T (Evans et al. 1998), to generate first strand cDNA. Using gene-specific nested primers and EPB, the 3′ end of the mRNA was amplified and TA-cloned for sequencing. The sequences were edited and assembled to obtain the complete sequence of TLGR-1. Forward and reverse primers were designed at the two ends to amplify the full-length cDNA, which was TA-cloned and sequenced in both directions using standard protocols.
TLGR-1 gene and promoter sequences
The complete genomic sequence of the TLGR-1 gene and its promoter elements were obtained by sequencing a cosmid clone containing the TLGR-1 gene. The tilapia genomic DNA library was constructed in SuperCos-1, as reported for channel catfish (Jaso-Friedmann et al. 2004). A directed polymerase chain reaction (PCR)-based iterative screening protocol was used to identify clones with TLGR-1 gene (Heaton et al. 1997). Specific PCR primers (TLGRCOSF1 5′-gcacccacgatatgacaaagttga-3′ and TLGRCOSR1 5′-ggcctttcaattttctcttacagaca-3′) were designed based on the cDNA sequence. After several rounds of screening, several TLGR-1-positive clones were isolated, expanded, and frozen in glycerol stocks for further analysis. The complete TLGR-1 gene sequence was obtained by sequencing the cosmid clone in a 373 A DNA sequencer (Applied Biosystems, CA) at the Integrated Biotech Laboratories (University of Georgia, Athens). The upstream region of the TLGR-1 gene (promoter region: 1,200 bp) was directly sequenced from the cosmid. Primers were synthesized as needed for “primer walking” (Genosys, TX). The TRANSFAC database (Wingender et al. 2000) and MatInspector web-based software (Quandt et al. 1995) were used to locate transcription factor binding sites.
Phylogenetic analysis and molecular modeling
Phylogenetic analysis and molecular modeling of TLGR-1 were done as described before (Praveen et al. 2004). Briefly, all the known granzymes and similar serine proteases were aligned, and phylogenetic analysis was done with Mega version 2.1 (Kumar et al. 2001). Analysis using different methods resulted in similar trees, and reliability of the trees was tested using 1,000 bootstrap replications. The tree was rooted on a subtree consisting of adipsin-like proteases from various sources. Molecular modeling of TLGR-1 was done using SWISS-MODEL in the first approach mode accessible via the Internet (http://www.expasy.org/swissmod). The crystal structure of human pro-granzyme K (1MZA) was used as a template to build the TLGR-1 model. RasWin version 2.6 was used to visualize the coordinate data.
Real-time polymerase chain reaction analysis
After exposing NCC to activation treatments, total RNA was isolated using RNeasy mini-kit (Qiagen, CA) following the manufacturer’s protocol, quantified spectrophotometrically, and treated with RNAse free DNAse (Promega, WI). The first strand cDNA was synthesized from 3 μg of total RNA using First Strand cDNA synthesis kit (Invitrogen). A cDNA stock for constructing a relative standard curve was synthesized using the same method. Primers were designed based on available gene sequences to amplify ∼150 bp amplicons for TLGR-1 and β-actin. Forward and reverse primers were selected at positions to include an intron so that genomic DNA contamination in the samples could be easily detected. A relative standard curve was constructed for target gene and housekeeping gene (β-actin) using either cDNA stock or full-length cDNA clone for TLGR-1 and β-actin. Briefly, reactions were set up with different concentrations ranging from 10 to 3000 pg per reaction for the standard curve. The reactions were carried out using Brilliant SYBR Green QPCR master mix (Stratagene, CA) according to manufacturer’s instructions. The efficiencies of the primer pairs were determined, and appropriate adjustments were made to optimize reaction conditions. Diluted cDNA samples were used as a template for every 25-μl reaction. Each sample was set up in triplicates for the PCR. The following PCR protocol was used for gene amplification: 95°C, 3 min; 40 cycles: denature 95°C, 30 s; anneal 60°C, 30 s; extend 72°C, 30 s, followed by 4°C hold using Mx3000P real-time PCR (RT-PCR) system (Stratagene).
Production of mature and pro-TLGR-1 in Pichia pastoris
The cDNAs coding for pro and mature forms of TLGR-1 were PCR-amplified and cloned in to pPICZ-alpha vector for expression in GS115 strain of P. pastoris (Invitrogen). The coding region of TLGR-1 contained a recognition site for XhoI. Because digestion of DNA with SalI has been shown to generate cohesive ends compatible with that made with XhoI digestion, forward primers TLGRYSTF1 (5′-aactgtcgacaaaagaatcataaatggca-3′) and TLGRYSTF2 (5′-aactgtcgacaaaagaagtgaaatcataaatggca-3′) were designed to engineer an SalI site upstream of mature and pro TLGR-1, respectively. A reverse primer TLGRYSTR1 (5′-ggggttctagatggcattgctttttattgagaatgt-3′) was designed to incorporate an XbaI site. The combination of these primers was used to PCR amplify the insert using a full-length TLGR-1 clone. The PCR products were purified, digested with a combination of restriction enzymes (XhoI and SalI), and ligated in to the pPICZ-alpha vector digested with XhoI and XbaI. The insert was introduced into this vector downstream of alpha factor signal sequence and Kex2 protease cleavage site. This position facilitated the production of recombinant proteins with native N-terminus and a polyhistidine tag at the C-terminus, which would be secreted to the medium with minimum contamination from yeast proteins. The ligation reaction was used to initially transform competent JM109 Escherichia coli cells, and positive clones were selected on a low-salt Luria–Bertani plate with 25 μg/ml Zeocin (Invitrogen). The clones were verified for the presence of insert by restriction digestion and colony PCR methods. Recombinant plasmids (pPICZ-alpha–TLGR-1 and pPICZ-alpha–pro-TLGR-1) were sequenced in both directions using 5′ and 3′ AOX1 primers (Invitrogen) to verify the correct reading frame. Subsequently, the recombinant plasmids were linearized by digesting with SacI before electroporating in to the GS115 strain of P. pastoris according manufacturer’s instructions. Positive clones were selected on yeast peptone dextrose (YPD) agar plates containing 100 μg/ml Zeocin. Positive clones were tested for insert by PCR. Expression of recombinant proteins was assessed by a small-scale expression trial followed by Western blot analysis of the trichloroacetic acid (TCA)-precipitated supernatants. Clones with high level of expression were selected to scale-up production.
To obtain enough recombinant protein for purification, the selected clones were grown in baffled flasks for optimum aeration. Using a single colony, 25 ml of buffered glycerol-complex medium [BMGY: 1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% yeast nitrogen base, 4×10−5% biotin, 1% glycerol] in a 250-ml baffled flask was incubated at 30°C for 18 h until the OD600=2–6. Cells were harvested by centrifugation, and the pellet was resuspended in buffered methanol-complex medium (BMMY: BMGY with glycerol replaced with 0.5% methanol) to an OD600 of 1.0 in a 1-l baffled flask. The flasks were incubated at 30°C for various time points until the optimum production was verified by Western blotting of supernatants collected at different time points. The supernatants were collected and stored at −80°C until ready for purification.
For purification of the His-tagged recombinant proteins, the supernatants were concentrated by ammonium sulfate precipitation followed by dialysis in phosphate-buffered saline (PBS, pH 7.2). The recombinant proteins were purified by metal affinity chromatography using Ni-NTA agarose (Qiagen) according to manufacturer’s instructions. The positive fractions were pooled, desalted, and concentrated using Centricon YM-3 centrifugal filters (Millipore, MA). Aliquots were stored at −80°C. Protein concentrations were estimated using Biorad Protein Assay Kit (Biorad, CA).
Western blot analysis
Proteins were resolved on 12.5% sodium dodecyl sulfate (SDS) gels, transferred to nitrocellulose membrane, and probed with INDIA-His probe–horse radish peroxidase (HRP) (Pierce, IL) or anti-His monoclonal antibody (Qiagen). Proteins were detected with SuperSignal West Pico Chemiluminescent Substrate (Pierce).
In vitro enzyme assays
Activity assays for proteases were performed at 37°C in 50 mM Tris–HCl, 0.15 M NaCl, 0.01% Triton X-100 (pH 7.6) containing 0.2 mM Ellman’s reagent [5,5′-dithiobis(2-nitrobenzoic acid), Sigma, MO] in 96-well microtiter plates with a reaction volume of 200 μl. Various thiobenzyl ester substrates at a concentration of 0.2 mM were used to determine the enzyme kinetics by measuring an increase in absorbance at the 405-nm wavelength over time with a Spectra Max Plus microplate reader (Molecular Devices, CA). Results for controls and samples are expressed as change in mOD/min as described before (Wilharm et al. 1999a). Stock solutions of Z-Lys-SBzl (Calbiochem, CA) were made in ethanol, while Z-Arg-SBzl, Boc-Ala-Ala-Asp-SBzl, Boc-Ala-Ala-Met-SBzl (MP Biomedicals, OH), N-Succinyl-Phe-Leu-Phe-SBzl, and N-Succinyl-Ala-Ala-Pro-Phe-pNA (Sigma) were dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C and added to the reaction buffer prior to the assay.
Results
Identification and analysis of full-length cDNA and gene for TLGR-1
For identification of genes involved in cytotoxic functions of NCC, a cDNA library was constructed using activated cytotoxic cells. The identification of a partial sequence of a serine protease with novel predicted substrate specificity from a tilapia NCC library has been reported (Praveen et al. 2004). A full-length cDNA for that protease was cloned from a cDNA library. Primers were designed based on this sequence to screen a tilapia genomic library, and a cosmid clone containing the gene and promoter elements was identified (Fig. 1). Surprisingly, the TLGR-1 gene had an approximate size of 1.32 kb (starting from the 5′ UTR and ending at the polyadenylation signal), which is much smaller than mammalian granzyme genes. However, the comparison of the cDNA and genomic sequences of the TLGR-1 gene revealed a five-exon and four-intron organization, which is shared among many members of the granzyme family. The teleost granzyme gene has shorter introns than the mammalian granzyme genes. All of the intron/exon boundaries of TLGR-1 gene fulfill the GT-AG rule (Mount 1982).


Genomic organization of TLGR-1 gene and promoter region. Exon sequences are represented in upper case letters, while introns and promoter region are represented as lower case letters. Putative transcription factor binding sites in the promoter region are represented as underlined bold letters with the name of the factor above the sequence. The predicted signal sequence at the N-terminus is highlighted, while the dipeptide is represented with double underline. A putative glycosylation site is boxed, and the polyadenylation signal is represented by underlined bold letters in the 3′ UTR. Triad residues constituting the active site are represented as bold letters in the sequence of amino acid chain
The proximal promoter region of TLGR-1 was analyzed for the putative transcription factor binding sites. Similar to other granzyme gene promoters, TLGR-1 has many binding sites for transcription factors specific for the immune system (Fig. 1). There are two AP-1 and NFAT binding sites along with sites for binding of interferon response factors and STATs. This would appear to suggest that there may be a significant correlation between the transcriptional upregulation of TLGR-1 and activated immune response reactions.
The TLGR-1 cDNA sequence was initially obtained by RT-PCR using specific primers at the 5′ and 3′ end of the gene, and the sequence was verified by comparing it with the genomic sequence. The cDNA and genomic sequences were submitted to GenBank (accession numbers AY918866 and AY918867, respectively). This cDNA has a total length of 999 bp, with an open reading frame of 765 bp that encodes a putative protein with 254 amino acids (Fig. 1). Using algorithms to predict signal sequences, a stretch of 23 residues at the N-terminal end was identified as a signal sequence, indicating that this protein is sorted to the secretory pathway. Signal sequence cleavage sites were predicted using weight matrix analysis of von Heijne (1986). TLGR-1 is predicted to have a dipeptide (Ser-Glu) as pro-peptide, much similar to the majority of mammalian granzymes. There is a single putative glycosylation site (NGTA) located toward the C-terminus of the protein (Fig. 1). The predicted molecular mass of the non-glycosylated mature TLGR-1 was calculated as 25,339 Da (229 amino acids) with an isoelectric point of 9.57. Other granzymes also have a highly basic charge.
Disulfide linkages are crucial for the proper folding of fully active granzymes. Overall structure of the tilapia granzyme had a close similarity to available crystal structures of mammalian granzymes. TLGR-1 can be predicted to have four disulfide linkages (Fig. 2), similar to what was shown for catfish granzyme (Praveen et al. 2004). Formation of disulfide bonds between Cys residues can be predicted as follows: 42 with 58, 136 with 168, 182 with 201. Cys191 and Cys220 (chymotrypsinogen numbering) could be analogous to a fourth disulfide linkage which bridges the active site serine, as in chymotrypsin. Although the tilapia granzyme has additional Cys residues (Fig. 2), these are not present at corresponding positions to that of granzyme A, which is crucial in the dimerization (Hink-Schauer et al. 2003).

Predicted three-dimensional model for TLGR-1. a Comparison of TLGR-1 structure to human granzyme K (1MZA.pdb). b Predicted disulfide bonds in TLGR-1. Cys residues are represented in green. Four disulfide bonds which are conserved in other granzyme sequences are represented by circles. The extra Cys residue is marked with an arrow (one additional Cys residue at the C-terminus of the molecule is omitted from the model and not shown)
Multiple sequence comparisons of TLGR-1 with related granzyme sequences enabled the identification of signature motifs shared by members of the granzyme family. While the N-terminal IIXG motif is conserved in the TLGR-1, the PHSRPYMA motif is not entirely conserved (Fig. 3). However, the residues in corresponding position share close similarity to those in the mammalian sequences. The three key amino acid residues representing the catalytic triad (charge relay system) of serine proteases (His57, Asp102, and Ser195; chymotrypsinogen numbering system) as well as their neighboring residues are well conserved in the TLGR-1 (Fig. 3). The combination of amino acid residues, which is crucial in the formation of the substrate specificity pocket, can be used as a prediction tool for primary specificity for a protease (Wouters et al. 2003). Based on the comparison of the residues in the TLGR-1, alignment with other granzyme sequences, and manual editing of the alignment based on crystallography structure profile (Hink-Schauer et al. 2002), a novel substrate specificity pocket triplet (GNN) can be predicted for this teleost granzyme. None of the other granzymes described to date share a specificity pocket triplet with TLGR-1. TLGR-1 has sequence similarity with almost all of the mammalian granzymes, with highest sequence similarity to granzyme B and G sequences.

Comparison of TLGR-1 with other granzyme sequences. Multiple sequence alignments showing the conserved regions in TLGR-1 sequence, which represent the signature motifs for granzyme-like proteases. Conserved residues at the catalytic triad are represented with a black circle above the alignment. Darker shading represents identical or residues with similar properties in 100% of the sequences, and lighter shading represents identical or residues with similar properties in 80% or more of all the sequences
Phylogenetic analysis of TLGR-1
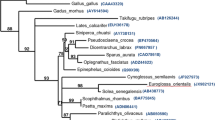
The TLGR-1 sequence was compared with known serine proteases, and the data were analyzed by different methods to obtain clues on the evolutionary position of this granzyme. Phylogenetic analysis using different methods yielded trees with a similar topology, and a representative tree is depicted in Fig. 4. To resolve the difference between closely related granzyme sequences, the tree was rooted on a subtree comprising of complement factor D, which has been shown to be evolutionarily distinct from granzymes while sharing many sequence features. TLGR-1 did not cluster with any of the mammalian granzymes. However, it grouped with other teleost granzymes identified by Expressed Sequence Tags (EST) database searches. Comparison of substrate specificity pocket triplet residues in the teleost sequences revealed a close similarity to TLGR-1, suggesting that they may share similar primary specificities.

Phylogenetic analysis of TLGR-1. Phylogram represents the evolutionary position of TLGR-1 compared to related serine proteases. The tree was derived by parsimony analysis, with Mega version 2.1. Sequences of mature protease were used in the analysis, and accession numbers are provided after the name. Numbers shown above the branches are bootstrap values based upon 1,000 replicates for parsimony. The tree was rooted on a subtree containing complement factor D to determine the clustering of TLGR-1 with similar granzyme
Expression pattern of TLGR-1
The expression of the TLGR-1 gene in various tissues was assessed by RT-PCR analysis using specific primers. Purified NCC were shown to be a major source for the expression of this gene (Fig. 5). Tissues rich in NCC [peripheral blood leukocytes (PBL) and anterior kidney] were the only places where the expression could be detected. Compared with the granzymes from catfish NCC, constitutive expression of TLGR-1 was found to be very low based on the intensity of the amplified bands.

Analysis of tissue expression of TLGR-1. RT-PCR analysis for TLGR-1 using cDNA from various tilapia tissues. Lanes: 1 purified NCC from peripheral blood, 2 total peripheral blood leukocytes, 3 total blood, 4 muscle, 5 liver, 6 gill, 7 spleen, 8 kidney, 9 TMB8. (A tilapia continuous cell line TMB-8 was provided by Dr. R. Hedrick, University of California-Davis. These cells were originally established from Oreochromis mossambicus cardiac tissue.) Beta actin was used as a normalizer
Transcriptional regulation of TLGR-1
The inducibility of the TLGR-1 message in tilapia NCC was assessed by RT-PCR. NCC were treated with various stimuli, and cDNA were prepared for analysis. Recombinant tilapia tumor necrosis factor (TNF) was found to have a profound effect on the transcription of TLGR-1 (Fig. 6a). Within 2 h of treatment with 100 ng/ml recombinant tilapia TNF, there was a 29-fold increase in TLGR-1 expression. The expression was reduced after 2 h but remained higher than base levels even after 4 h. Similarly, treatment of NCC with phorbol 12-myristate-13-acetate (PMA)/calcium ionophores resulted in more than twofold increase in TLGR-1 expression (Fig. 6b). More than sixfold increase in expression could be observed by treating the cells with lipopolysaccharide (LPS) for 4 h (Fig. 6c).

Transcriptional regulation of TLGR-1 in NCC. RT-PCR analysis of cDNA from NCC purified from tilapia peripheral blood after subjecting to different treatments. a NCC were treated with recombinant tilapia TNF-alpha and incubated for 2 or 4 h and analyzed for TLGR-1 expression. b NCC treated with PMA and calcium ionophore (A23187) for 2 or 4 h. c NCC treated with lipopolysaccharides (from E. coli)
Expression of recombinant TLGR-1
To express the pro and mature forms of TLGR-1, expression cassettes were engineered so that it could be inserted between XhoI and XbaI sites of yeast expression vector, pPICZ-alpha. Recombinant vectors were introduced in to the GS115 strain of P. pastoris so that the recombinant proteins were expressed as a fusion protein with an N-terminal alpha signal sequence and C-terminal granzyme. By inserting a Kex2 recognition sequence in between the signal sequence and granzyme, active (cleaved) TLGR-1 could be secreted in to the culture supernatant. Incorporation of small C-terminal tags without affecting the protease activity has been demonstrated for many proteases before (Beresford et al. 1997; Huang et al. 1997 1998; Xia et al. 1998). Both pro and mature recombinant TLGR-1 had a c-myc tag followed by a 6x-His-tag at the C-terminus (Fig. 7a). Expression of pro-TLGR-1 was achieved by retaining the activation dipeptide at the N-terminus. The dipeptide is known to yield inactive enzyme due to improper protein folding.

Production of recombinant TLGR-1 in P. pastoris. a Representation of expression cassettes engineered to generate pro and mature TLGR-1 using pPICZ-alpha expression vector. Pro and mature forms of TLGR-1 were expressed as fusion proteins starting with the yeast alpha factor at the N-terminus and polyhistidine and c-myc tagged granzyme at the C-terminus. Presence of a Kex2 site in between the alpha factor and the granzyme allowed the expression of granzyme with a native N-terminus, which can be secreted in to the culture medium with minimum contaminating yeast proteins. b Optimization of recombinant protein expression. The culture supernatants were collected at the time points indicated and subjected to TCA precipitation followed by Western blotting. His-tagged proteins were detected with INDIA-His probe conjugated to HRP followed by chemiluminescence. Yeast transfected with empty vector alone (E) also were checked for the expression of proteins, and only the 72 h time point is shown. The predicted bands representing TLGR and pro-TLGR are highlighted with arrowheads
Several expression clones were identified by plating the transformed cells on YPD plates with Zeocin. Small-scale expression trials were done to assess the level of expression of selected clones upon induction with methanol (data not shown). Clones with highest expression levels were selected for further studies. Single yeast colony was grown and induced to secrete the recombinant proteins in 300 ml culture volume in 1,000-ml baffled flasks, and culture supernatants were collected at various time points for TCA precipitation. The proteins were resolved on a 12.5% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose. Expression levels of recombinant proteins were assessed by staining with His probe. A protein of approximately 33 kDa was found to be upregulated in both mature and pro TLGR-1 culture supernatants (Fig. 7b). For mature TLGR-1, a high level of expression of recombinant proteins was observed from 24 h lasting until 72 h. From that point, there was significant degradation of the proteins. Similarly, expression levels were high for pro-TLGR-1 from 48 h and remained high for 96 h (Fig. 7b).
High level expression and purification of recombinant TLGR-1
To assess the success of metal affinity chromatography, Ni-NTA agarose beads were mixed with a small volume (20 ml) of the culture supernatants, and His-tagged proteins were purified eluting the bound proteins in a buffer containing 250 mM imidazole. Almost all of the recombinant proteins were eluted in the first elutions, detected by Western blotting and protease activity assays (data not shown). Later, large volumes (1–2 l) of the culture supernatants were subjected to ammonium sulfate precipitation to concentrate the proteins. This was followed by nickel affinity chromatography to purify His-tagged proteins (Fig. 8).

Purification of pro and mature TLGR-1 recombinant proteins. Supernatants were collected after methanol induction for 72 h, and total proteins were concentrated by ammonium sulfate precipitation. His-tagged proteins were purified by nickel affinity chromatography. a Purification success of recombinant proteins verified by Western blotting and detection with INDIA-His probe. Lanes: V vector alone; F flow through from the column; E1, E2, and E3 three consecutive elutions of the column using buffer containing 250 mM imidazole. b Purity of the protein preparations verified by Coomassie staining of the gel. Lanes: 1 Pro-TLGR, 2 TLGR
In vitro esterolytic activity of recombinant TLGR-1
Due to the novel specificity pocket triplet residues in TLGR-1, it was expected not to hydrolyze thiobenzyl esters specially designed for granzymes with tryptase, Met-ase, or Asp-ase activity. The hydrolysis of synthetic chymase substrates was used to analyze the activity of TLGR-1. Effective hydrolysis of N-Succinyl-Phe-Leu-Phe-SBzl (Fig. 9a) and N-Succinyl-Ala-Ala-Pro-Phe-pNA (Fig. 9b) by TLGR-1 suggested its strong preference for bulky residues at the P1 site. Later, a comparison of the hydrolysis of various thiobenzyl ester substrates revealed in fact that TPCR-1 had chymase activity TLGR-1 (Fig. 10). The pro-TLGR-1 failed to cleave any of the substrates, indicating that the presence of pro-peptide at the N-terminus prevented proper folding of the protease, which is crucial for its activity.

Enzymatic activity of recombinant TLGR-1. a Plot depicting the change in optical density against time from an in vitro enzyme kinetic assay using N-succinyl-Phe-Leu-Phe-SBzl as a substrate. b Hydrolysis of N-succinyl-Ala-Ala-Pro-Phe-pNA by mature and pro-TLGR-1. Both the substrates were used at a final concentration of 0.3 mM, and enzyme concentration was approximately 1 nM for both mature and pro-TLGR-1

Substrate specificity of TLGR-1. The enzymatic activity of mature and pro-TLGR-1 was measured using the indicated thiobenzyl ester substrates in a final concentration of 0.3 mM. Enzyme concentration was approximately 1 nM for both mature and pro-TLGR-1. Change in optical density was measured for 30 min, and rate of hydrolysis is depicted as change in mOD/min
Discussion
We have previously reported the expression of granzyme-like serine proteases in NCC purified from the peripheral blood of tilapia (Praveen et al. 2004). The full-length cDNA coding for TLGR-1 was identified and sequenced in two directions to obtain the putative amino acid sequence of the protein and to identify sequence motifs shared with other granzymes. Later, a genomic library was screened using specific primers to obtain a cosmid clone containing full-length TLGR-1 gene. We sequenced genomic DNA comprising the full gene and the promoter region (Fig. 1). Compared to other granzyme sequences, the TLGR-1 gene had a smaller size. However, the intron–exon structure of the gene had closest similarity to other mammalian granzymes (Kelly et al. 1996; Trapani 2001). Additionally, the number and position of introns of TLGR-1 suggest that this could be a member of the sixth class of serine proteases (Irwin et al. 1988), like NK cell-specific granzyme M (Kelly et al. 1996; Pilat et al. 1994). The first one of the substrate specificity triplet residues (Gly 173; −6 with reference to active site Ser) is encoded within the fourth exon (Fig. 1), as in the case of other chymotrypsin-like proteases (Bell et al. 1984).
Studies on proximal promoter elements of mammalian granzymes had revealed the conserved sequences enabling cell-specific expression of these proteases. Granzyme B promoter has binding sites for T cell-specific transcription factors such as Ikaros and core-binding factor as well as for more ubiquitous transcription factors like AP-1 (Haddad et al. 1993; Kamachi et al. 1990; Wang and Speck 1992). These sequences are found to be sufficient to drive reporter gene expression in T cell lines where these factors are constitutively active (Fregeau and Bleackley 1991; Hanson et al. 1991). The promoter region of granzyme M gene has been shown to have binding sites for factors like AP-2, AP-3, and glucagons-G3A, influencing its bias for expression in NK cells (Kelly et al. 1996; Smyth et al. 1995a). The presence of multiple transcription factors in the promoter region of the TLGR-1 gene in NCC is an indirect indication that this gene is highly regulated during the immune response.
Highly conserved Cys residues at similar positions in all granzyme sequences give rise to three pairs of disulfide bonds, which are crucial in proper folding and activation of granzyme during processing. Among these disulfide bonds, the ones between Cys26 and Cys42 as well as between Cys151 and Cys167 link the primary and secondary substrate-binding sites, forming S1 subsites in granzymes A, K, and M (Sattar et al. 2003). A fourth pair of Cys residues forms an additional disulfide bond, which bridges the active site Ser residue is found in granzymes M and K (Kelly et al. 1996; Przetak et al. 1995). Granzyme A is the only mammalian granzyme shown to have one additional Cys residue, which is responsible for intermolecular disulfide bonds (Bell et al. 2003; Hink-Schauer et al. 2003). TLGR-1 was shown to have two additional Cys residues (Fig. 2). At this time, their function is unknown, as their positions do not match with those corresponding to the dimer-forming Cys residue of granzyme A (Hink-Schauer et al. 2003).
Multiple sequence alignment of TLGR-1 with other granzymes revealed highly conserved regions shared by all known granzymes (Fig. 3). The N-terminal IIXG motif and catalytic triad residues that make up the charge relay system were highly conserved. Moreover, residues around the triad residues were highly conserved among all the sequences. The presence of Gly residue at position −6 upstream of the active site Ser indicated a substrate specificity that was different from that of granzymes A/K, B, and M. Other residues at positions +15 to +17 and +28 relative to the active site Ser are found to be important in determining the primary substrate specificity of Ser protease (Bode et al. 1989). TLGR-1 has Ser-Phe-Asn (+15 to +17) and Asn (+28) at these positions, which is quite different from all other known granzymes. A comparison of crystal structures of Ser proteases has led to a theory that three residues of the S1 substrate-binding pocket confer primary specificity. These specificity-conferring residues are located at positions 189, 216, and 226 (chymotrypsin numbering; Wouters et al. 2003). Aligning the TLGR-1 sequence with other granzymes and matching the secondary structure elements, the substrate specificity pocket residues for this protease were determined as Gly-Asn-Asn. These triplet amino acid residues have not been previously described in mammalian granzymes.
Multiple sequence comparisons alone are not sufficient to draw conclusions on the evolutionary positions of closely related proteins like granzymes. Phylogenetic analysis of mammalian granzymes has revealed the clustering of granzymes with similar substrate specificity, giving rise to the classification of granzymes into three major groups (Grossman et al. 2003; Wouters et al. 2003). Similar analysis of teleost granzymes along with mammalian serine protease placed TLGR-1 in a separate cluster, indicating a parallel evolution for teleost granzymes. Catfish granzymes with similar predicted substrate specificity were the other members of that cluster. The experimental verification of the enzymatic activity for the catfish granzymes would be crucial to make the correlation between substrate specificity and sequence similarity.
The pattern of tissue expression of TLGR-1 suggests that this protease is associated with cytotoxic cell populations (Fig. 5). Tissues with appreciable TLGR-1 expression were the ones with high NCC density. The use of cDNA derived from total tissues for expression studies could be misleading due to the interference from different cell populations that might be present in these tissues. The cell line (TMB-8), which has been derived from heart tissue, was used to assess the specificity of expression with more confidence. TMB-8 cells have been used as nonhematopoietic and noncytotoxic controls in previous experiments (Taylor et al. 2001). Compared to the CFGR-1, which had a high level of constitutive expression in NCC from different tissues (Praveen et al. 2004), TLGR-1 expression levels were very low in resting NCC (Fig. 5). Transcriptional upregulation of TLGR-1 with various stimuli indicated the high degree of inducibility for this tilapia granzyme (Fig. 6). Transcriptional activation of granzyme A in response to TNF has been previously reported in lymphokine-activated killer cells and CTLs (Owen-Schaub et al. 1989; Robinet et al. 1990).
Purification of catalytically active recombinant granzymes is critical in understanding their individual physiological roles, compared to combined effects of multiple granzymes in purified cytotoxic granules. The generation of mammalian granzymes in recombinant forms has been reported (Beresford et al. 1997; Kummer et al. 1996; Pham et al. 1998; Smyth et al. 1995b; Sun et al. 1999; Wilharm et al. 1999a; Xia et al. 1998). The use of the P. pastoris expression system with pPICZ-alpha expression vector allowed the generation of recombinant granzymes with the desired N-terminal end. By inserting the tilapia granzyme expression sequence downstream of an alpha mating signal sequence, secretion of the protein to the expression medium was possible. The pro-TLGR-1 was generated by retaining two residues at the N-terminus of the mature protease (Fig. 7a). This has been shown to prevent the proper folding and activation of mammalian recombinant granzymes (Kummer et al. 1996; Sayers et al. 1996; Sun et al. 1999). The supernatants were collected at various time points, and expression levels were compared by Western blotting (Fig. 7b).
Purification of recombinant TLGR-1 from culture supernatants was carried out after concentrating the proteins by ammonium sulfate precipitation. This method had been previously used without affecting the proteolytic activities of human mast cell chymase (Lockhart et al. 2005). After the ammonium sulfate precipitation, the pellet was dissolved in PBS and concentrated to 50 ml followed by dialyzing against PBS to remove traces of ammonium sulfate. His-tagged proteins were purified by nickel affinity chromatography (Fig. 8). The Western blot analysis of TLGR-1 suggests that the protein might be glycosylated. The predicated molecular mass of mature TLGR-1 is 25.3 kDa, while the protein appeared to have an approximate mass of 33 kDa, suggesting the presence of sugar residues in the predicted glycosylation site on the polypeptide chain. Glycosylation of granzymes is critical, because they have been shown to be targeted to the lytic granules by the mannose-6-phosphate receptor (Griffiths and Isaaz 1993).
Granzymes are characterized by their very narrow substrate specificity. The requirement for P1 residue varies significantly between different classes of mammalian granzymes (Kam et al. 2000). In most cases, the substrate specificity of individual granzymes can be predicted based on the architecture of its substrate-binding pocket (Wouters et al. 2003). Such prediction algorithms suggested that TLGR-1 had a preference for bulkier P1 residues than that for tryptases, Asp-ases, or Met-ases. Using synthetic substrates designed with a bulky Phe residue at the P1 site, the activity of TLGR-1 was shown to be similar to that of mammalian chymases (Figs. 9 and 10).
In conclusion, the novel serine protease identified from tilapia NCC was shown to have chymase activity. It is not clear whether these cells do express other granzymes and other components of granule exocytosis pathway. The TLGR-1 gene is highly inducible with various stimuli. Compared to the expression pattern of similar granzymes in channel catfish NCC, tilapia granzymes require longer activation for the transcription and translation of cytotoxic molecules, as determined by the protease activity of supernatants from granule exocytosis assay (data not shown). Studies are underway to uncover the physiological roles of this protease in these cytotoxic cells and also to identify other granule components in tilapia NCC.
References
Bell GI, Quinto C, Quiroga M, Valenzuela P, Craik CS, Rutter WJ (1984) Isolation and sequence of a rat chymotrypsin B gene. J Biol Chem 259:14265–14270
Bell JK, Goetz DH, Mahrus S, Harris JL, Fletterick RJ, Craik CS (2003) The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat Struct Biol 10:527–534
Beresford PJ, Kam CM, Powers JC, Lieberman J (1997) Recombinant human granzyme A binds to two putative HLA-associated proteins and cleaves one of them. Proc Natl Acad Sci U S A 94:9285–9290
Beresford PJ, Zhang D, Oh DY, Fan Z, Greer EL, Russo ML, Jaju M, Lieberman J (2001) Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem 276:43285–43293
Bode W, Meyer E Jr, Powers JC (1989) Human leukocyte and porcine pancreatic elastase: x-ray crystal structures, mechanism, substrate specificity, and mechanism-based inhibitors. Biochemistry 28:1951–1963
Carlson RL, Evans DL, Graves SS (1985) Nonspecific cytotoxic cells in fish (Ictalurus punctatus). V. Metabolic requirements of lysis. Dev Comp Immunol 9:271–280
Ebnet K, Hausmann M, Lehmann-Grube F, Mullbacher A, Kopf M, Lamers M, Simon MM (1995) Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J 14:4230–4239
Evans DL, Leary JH III, Jaso-Friedmann L (1998) Nonspecific cytotoxic cell receptor protein-1: a novel (predicted) type III membrane receptor on the teleost equivalent of natural killer cells recognizes conventional antigen. Cell Immunol 187:19–26
Faisal M, Ahmed II, Peters G, Cooper EL (1989) Natural cytotoxicity of tilapia leukocytes. Dis Aquat Org 7:17–22
Fan ZS, Beresford PJ, Zhang D, Lieberman J (2002) HMG2 interacts with the nucleosome assembly protein SET and is a target of the cytotoxic T-lymphocyte protease granzyme A. Mol Cell Biol 22:2810–2820
Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J (2003a) Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112:659–672
Fan ZS, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, Lieberman J (2003b) Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol 4:145–153
Fregeau CJ, Bleackley RC (1991) Transcription of two cytotoxic cell protease genes is under the control of different regulatory elements. Nucleic Acids Res 19:5583–5590
Graves SS, Evans DL, Dawe DL (1985) Antiprotozoan activity of nonspecific cytotoxic cells (NCC) from the channel catfish (Ictalurus punctatus). J Immunol 134:78–85
Greenlee AR, Brown RA, Ristow SS (1991) Nonspecific cytotoxic cells of rainbow trout (Oncorhynchus mykiss) kill YAC-1 targets by both necrotic and apoptic mechanisms. Dev Comp Immunol 15:153–164
Griffiths GM, Isaaz S (1993) Granzymes A and B are targeted to the lytic granules of lymphocytes by the mannose-6-phosphate receptor. J Cell Biol 120:885–896
Grossman WJ, Revell PA, Lu ZH, Johnson H, Bredemeyer AJ, Ley TJ (2003) The orphan granzymes of humans and mice. Curr Opin Immunol 15:544–552
Haddad P, Wargnier A, Bourge JF, Sasportes M, Paul P (1993) A promoter element of the human serine esterase granzyme B gene controls specific transcription in activated T cells. Eur J Immunol 23:625–629
Han J, Goldstein LA, Gastman BR, Froelich CJ, Yin XM, Rabinowich H (2004) Degradation of MCL-1 by granzyme B: implications for bim-mediated mitochondrial apoptotic events. J Biol Chem 279:22020–22029
Hanson RD, Sclar GM, Kanagawa O, Ley TJ (1991) The 5'-flanking region of the human CGL-1/granzyme B gene targets expression of a reporter gene to activated T-lymphocytes in transgenic mice. J Biol Chem 266:24433–24438
Heaton MP, Lopez-Corrales NL, Smith TPL, Kappes SM, Beattie CW (1997) Directed cosmid isolation of bovine markers for physical assignment by fish. Anim Biotechnol 8:167–177
Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ (1994) Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 76:977–987
Hink-Schauer C, Estebanez-Perpina E, Wilharm E, Fuentes-Prior P, Klinkert W, Bode W, Jenne DE (2002) The 2.2-A crystal structure of human pro-granzyme K reveals a rigid zymogen with unusual features. J Biol Chem 277:50923–50933
Hink-Schauer C, Estebanez-Perpina E, Kurschus FC, Bode W, Jenne DE (2003) Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat Struct Biol 10:535–540
Huang C, Wong GW, Ghildyal N, Gurish MF, Sali A, Matsumoto R, Qiu WT, Stevens RL (1997) The tryptase, mouse mast cell protease 7, exhibits anticoagulant activity in vivo and in vitro due to its ability to degrade fibrinogen in the presence of the diverse array of protease inhibitors in plasma. J Biol Chem 272:31885–31893
Huang C, Friend DS, Qiu WT, Wong GW, Morales G, Hunt J, Stevens RL (1998) Induction of a selective and persistent extravasation of neutrophils into the peritoneal cavity by tryptase mouse mast cell protease 6. J Immunol 160:1910–1919
Irwin DM, Robertson KA, MacGillivray RT (1988) Structure and evolution of the bovine prothrombin gene. J Mol Biol 200:31–45
Jaso-Friedmann L, Evans DL (1999) Mechanisms of cellular cytotoxic innate resistance in tilapia (Oreochromis nilotica). Dev Comp Immunol 23:27–35
Jaso-Friedmann L, Harris DT, St. John A, Koren HS, Evans DL (1990) A monoclonal antibody-purified soluble target cell antigen inhibits nonspecific cytotoxic cell activity. J Immunol 144:2413–2418
Jaso-Friedmann L, Peterson DS, Gonzalez DS, Evans DL (2002) The antigen receptor (NCCRP-1) on catfish and zebrafish nonspecific cytotoxic cells belongs to a new gene family characterized by an F-box-associated domain. J Mol Evol 54:386–395
Jaso-Friedmann L, Praveen K, Leary JH III, Evans DL (2004) The gene and promoter structure of non-specific cytotoxic cell receptor protein-1 (NCCRP-1) in channel catfish (Ictalurus punctatus). Fish Shellfish Immunol 16:553–560
Kam CM, Hudig D, Powers JC (2000) Granzymes (lymphocyte serine proteases): characterization with natural and synthetic substrates and inhibitors. Biochim Biophys Acta 1477:307–323
Kamachi Y, Ogawa E, Asano M, Ishida S, Murakami Y, Satake M, Ito Y, Shigesada K (1990) Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol 64:4808–4819
Kelly JM, O’Connor MD, Hulett MD, Thia KY, Smyth MJ (1996) Cloning and expression of the recombinant mouse natural killer cell granzyme Met-ase-1. Immunogenetics 44:340–350
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Kummer JA, Kamp AM, Citarella F, Horrevoets AJ, Hack CE (1996) Expression of human recombinant granzyme A zymogen and its activation by the cysteine proteinase cathepsin C. J Biol Chem 271:9281–9286
Lieberman J (2003) The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol 3:361–370
Lieberman J, Fan Z (2003) Nuclear war: the granzyme A-bomb. Curr Opin Immunol 15:553–559
Lockhart BE, Vencill JR, Felix CM, Johnson DA (2005) Recombinant human mast-cell chymase: an improved procedure for expression in Pichia pastoris and purification of the highly active enzyme. Biotechnol Appl Biochem 41:89–95
McKinney EC, Schmale MC (1994) Damselfish with neurofibromatosis exhibit cytotoxicity toward tumor targets. Dev Comp Immunol 18:305–313
Mount SM (1982) A catalogue of splice junction sequences. Nucleic Acids Res 10:459–472
Owen-Schaub LB, Crump WL III, Morin GI, Grimm EA (1989) Regulation of lymphocyte tumor necrosis factor receptors by IL-2. J Immunol 143:2236–2241
Pham CT, Thomas DA, Mercer JD, Ley TJ (1998) Production of fully active recombinant murine granzyme B in yeast. J Biol Chem 273:1629–1633
Pilat D, Fink T, Obermaier-Skrobanek B, Zimmer M, Wekerle H, Lichter P, Jenne DE (1994) The human Met-ase gene (GZMM): structure, sequence, and close physical linkage to the serine protease gene cluster on 19p13.3. Genomics 24:445–450
Praveen K, Evans DL, Jaso-Friedmann L (2004) Evidence for the existence of granzyme-like serine proteases in teleost cytotoxic cells. J Mol Evol 58:449–459
Przetak MM, Yoast S, Schmidt BF (1995) Cloning of cDNA for human granzyme 3. FEBS Lett 364:268–271
Quandt K, Frech K, Karas H, Wingender E, Werner T (1995) MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res 23:4878–4884
Revell PA, Grossman WJ, Thomas DA, Cao X, Behl R, Ratner JA, Lu ZH, Ley TJ (2005) Granzyme B and the downstream granzymes C and/or F are important for cytotoxic lymphocyte functions. J Immunol 174:2124–2131
Robinet E, Branellec D, Termijtelen AM, Blay JY, Gay F, Chouaib S (1990) Evidence for tumor necrosis factor-alpha involvement in the optimal induction of class I allospecific cytotoxic T cells. J Immunol 144:4555–4561
Russell JH, Ley TJ (2002) Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 20:323–370
Sattar R, Ali SA, Abbasi A (2003) Bioinformatics of granzymes: sequence comparison and structural studies on granzyme family by homology modeling. Biochem Biophys Res Commun 308:726–735
Sayers TJ, Lloyd AR, McVicar DW, O’Connor MD, Kelly JM, Carter CR, Wiltrout TA, Wiltrout RH, Smyth MJ (1996) Cloning and expression of a second human natural killer cell granule tryptase, HNK-Tryp-2/granzyme 3. J Leukoc Biol 59:763–768
Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J (2005) Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J Exp Med 201:465–471
Smyth MJ, Hulett MD, Thia KY, Young HA, Sayers TJ, Carter CR, Trapani JA (1995a) Cloning and characterization of a novel NK cell-specific serine protease gene and its functional 5'-flanking sequences. Immunogenetics 42:101–111
Smyth MJ, O’Connor MD, Kelly JM, Ganesvaran P, Thia KY, Trapani JA (1995b) Expression of recombinant human Met-ase-1: a NK cell-specific granzyme. Biochem Biophys Res Commun 217:675–683
Sun J, Bird CH, Buzza MS, McKee KE, Whisstock JC, Bird PI (1999) Expression and purification of recombinant human granzyme B from Pichia pastoris. Biochem Biophys Res Commun 261:251–255
Suzumura E, Kurata O, Okamoto N, Ikeda Y (1994) Characteristics of natural killer-like cells in carp. Fish Pathol 29:199–203
Taylor SL, Jaso-Friedmann L, Allison AB, Eldar A, Evans DL (2001) Streptococcus iniae inhibition of apoptosis of nonspecific cytotoxic cells: a mechanism of activation of innate immunity in teleosts. Dis Aquat Org 46:15–21
Trapani JA (2001) Granzymes: a family of lymphocyte granule serine proteases. Genome Biol 2:3014.1–3014.7
Trapani JA, Smyth MJ (2002) Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2:735–747
Trapani JA, Sutton VR (2003) Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol 15:533–543
von Heijne G (1986) A new method for predicting signal sequence cleavage sites. Nucleic Acids Res 14:4683–4690
Wang SW, Speck NA (1992) Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol 12:89–102
Wilharm E, Parry MA, Friebel R, Tschesche H, Matschiner G, Sommerhoff CP, Jenne DE (1999a) Generation of catalytically active granzyme K from Escherichia coli inclusion bodies and identification of efficient granzyme K inhibitors in human plasma. J Biol Chem 274:27331–27337
Wilharm E, Tschopp J, Jenne DE (1999b) Biological activities of granzyme K are conserved in the mouse and account for residual Z-Lys-SBzl activity in granzyme A-deficient mice. FEBS Lett 459:139–142
Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I, Schacherer F (2000) TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res 28:316–319
Wouters MA, Liu K, Riek P, Husain A (2003) A despecialization step underlying evolution of a family of serine proteases. Mol Cell 12:343–354
Xia Z, Kam CM, Huang C, Powers JC, Mandle RJ, Stevens RL, Lieberman J (1998) Expression and purification of enzymatically active recombinant granzyme B in a baculovirus system. Biochem Biophys Res Commun 243:384–389
Acknowledgements
We thank Drs. Dorothy Hudig, Dieter Jenne, Jan Potempa, and James Powers for generously providing reagents and valuable suggestions.
This work was supported by BARD # US-3159-99C and Veterinary Medical Experimental Station Grants at the College of Veterinary Medicine, University of Georgia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Praveen, K., Leary, J.H., Evans, D.L. et al. Molecular characterization and expression of a granzyme of an ectothermic vertebrate with chymase-like activity expressed in the cytotoxic cells of Nile tilapia (Oreochromis niloticus) . Immunogenetics 58, 41–55 (2006). https://doi.org/10.1007/s00251-005-0063-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-005-0063-4