
Abstract
Transmission electron microscopy is a powerful technique for studying the three-dimensional (3D) structure of a wide range of biological specimens. Knowledge of this structure is crucial for fully understanding complex relationships among macromolecular complexes and organelles in living cells. In this paper, we present the principles and main application domains of 3D transmission electron microscopy in structural biology. Moreover, we survey current developments needed in this field, and discuss the close relationship of 3D transmission electron microscopy with other experimental techniques aimed at obtaining structural and dynamical information from the scale of whole living cells to atomic structure of macromolecular complexes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transmission electron microscopy is a powerful method to study the ultra-structure of cell components. This technique went through major improvements since the first commercial transmission electron microscope in 1939. These changes concern three main points:
-
Resolution improved dramatically with the development of more powerful microscopes based on the voltage increment and on more coherent emission sources.
-
New sample preparation techniques changed the setup of more preservative procedures for the observation of biological samples in conditions closer to live cells and protein complexes.
-
Development of computing techniques allowed the computation and analysis of massive data and access to three-dimensional (3D) information.
The access to 3D information constitutes the latest revolution in transmission electron microscopy and is changing the vision of cells by the development of a new concept named “integrative imaging” (Gue et al. 2005). This term refers to the possibility of combining protein structures at atomic resolution (from Nuclear Magnetic Resonance (NMR) and X-ray crystallography) with 3D reconstructions of macromolecular complexes at nanometric resolution (from 3D cryo-electron microscopy), and the possibility of integrating these structures within massive volumes of cellular organelles (from tomographic 3D transmission electron microscopy), or even within living or frozen-hydrated whole cells observed using fluorescence or confocal optical microscopy. Hence, integrative imaging is the forefront approach for studying complex and dynamic structure–function relationships of nanomachines within cells.
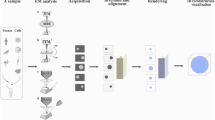
Depending on the nature of each sample, different strategies must be chosen (Fig. 1). Thus, if the sample contains a single object, electron tomography should be chosen. This is the case for most cellular organelles (mitochondria, Golgi, etc.). Electron tomography allows 3D reconstruction of a single object after acquisition of tilt series, corresponding to a set of 2D projections of this single object, with the sample gradually rotated along an axis defined as the tilt axis. Conversely, when the sample is composed of multiple copies of the same object, such as proteins and macromolecular complexes, the strategy of data collection and 3D reconstruction is different and aims at the computation of a 3D average map. Here again, different approaches can be taken, depending on internal symmetries of the particles and on the regular organization of the particles (low point-group symmetries, 2D crystals, icosahedral, or helical). Hence, 3D reconstruction of samples presenting helical or icosahedral symmetry can be theoretically performed from a single electron microscope image, after averaging in 3D all correctly oriented 2D projections of the objects, and by imposing correct point-group symmetries. When these particles lack any particular symmetry (as is the case of the ribosome), or show only low point-group symmetries (GroEL), an approach named “single particle analysis” can be applied, either using tilted pairs of images (45° and 0° tilt for random conical tilt, Radermacher 1988, and +45° and −45° for orthogonal reconstruction technique, Leschziner and Nogales 2006) or using 0°-tilt images and relying on sinograms (van Heel 1987). In the case of 2D crystals, numerous copies of the sample are regularly assembled on large lattices. In this case, even though a 3D average map is computed from many copies of the object, all particles are forced to be in a single orientation within the lattice plane. Therefore, the 3D reconstruction of 2D crystals requires tilting of the grid in the microscope to record tilt series for a 3D reconstruction based on tomographic principles.

Strategies for integrative 3D recontruction of biological samples
Transmission electron microscopy principles
The principle of transmission electron microscope is similar to that of a light microscope except that the illumination beam is formed by electrons instead of photons. Electrons are generated at the cathode (tungsten filament, LaB6 crystal, or field emission gun) and accelerated by a cascade of voltage differences (ranging from 80 kV to 1 MV depending on the microscope) with a nearby anode. The electron beam travels inside the microscope under high vacuum to avoid scattering due to a possible interaction of electrons with gas molecules. Several coils act as electromagnetic lenses and focus the beam on the sample. Assuming that the specimen is thin enough (weak-phase-objects, Frank 2006), the acquired images can be modeled as projection images of the specimen. Tilt of the specimen-holder produces different views that can be subsequently used for tomographic 3D reconstruction. The use of accelerated electrons implies very short wavelengths of the electron beam. If microscopes were totally devoid of defects, their resolution would only be limited by the wavelength of the beam; therefore, even at 80kV, the theoretical resolution limit that could be reached by an electron microscope should be close to 1/0.15 Å−1 (Nellist et al. 2004). However, this high resolution is never achieved (for measures of resolution of reconstructed volumes, see (Frank 2002; Frank et al. 1981; Harauz and van Heel 1986; Penczek 2002; Radermacher 1988; Rosenthal and Henderson 2003; Saxton and Baumeister 1982; Sousa and Grigorieff 2007; Unser et al. 1987, 2005; van Heel 1982; van Heel and Schatz 2005), especially with biological samples that impose additional limiting factors: radiation damage, sample heterogeneity, microscope aberrations, additive noise and missing orientations.
-
Radiation damage. Biological material is very sensitive to the radiation damage caused by the electron beam. Effects of the beam on the sample include heating, chemical degradation, bubbling and mass loss (Egerton et al. 2004; Glaeser 1971, 1999). The damage is particularly harmful for samples that are imaged more than once (for instance, when the same sample is tilted several times to acquire different projection views). Very low radiation dose must be used in this case to avoid specimen damage and preserve the structural integrity of biological material. Moreover, keeping samples at low temperature prevents diffusion of high-reactive free radicals generated by the electron beam and contribute to sample preservation. In general, acceptable cumulated radiation dose is below 30–50 electrons Å−2. For a low-dose electron microscopy, one usually uses doses that are kept below 10 electrons Å−2. The lower the dose, the more noisy and less contrasted are the images. This limitation can be compensated by taking into account more projections and by averaging. For instance, Unwin and Henderson reconstructed a map of bacteriorhodopsin at a resolution of 1/7Å−1 using a crystal of 10,000 unit cells and a dose of 0.5 electrons Å−2 (Unwin and Henderson 1975). Since a crystal has multiple copies of similar structures regularly ordered in space, one can average all projections and produce a much cleaner projection whose structural information has been preserved up to high resolution.
-
Structural heterogeneity. Since particles being imaged may be flexible, they can adopt different conformations to perform molecular functions. Hence, micrographs may contain biochemically identical particles with small structural differences (Brink et al. 2004; Gao et al. 2004; White et al. 2004). However, reconstruction algorithms assume that image sets correspond to homogeneous populations. Several methods have been developed to tackle structural heterogeneity: Normal mode analysis (Tama et al. 2004a, b), Multivariate statistical analysis (Frank 2006; van Heel 1984; van Heel and Frank 1981), Analysis of the 3D variance (Penczek et al. 2006a, b) and Maximum likelihood (Scheres et al. 2005b, 2007; Sigworth 1998). These methods study specimen variability either by separating projections in homogeneous populations, or explicitly considering and estimating the variability present in projections, or even by identifying those regions in space that are more likely to vary from one image to another. For an extensive review on the topic, see Leschziner and Nogales (2007).
-
Microscope aberrations. Electron microscopes slightly distort images of the observed object. One should consider spherical and chromatic aberrations, instabilities of magnetic lenses and instabilities of electron acceleration, etc. (Frank 2006). Image formation with all aberrations is modeled by the Contrast Transfer Function (CTF). The CTF is expressed in reciprocal space and its equivalent in real space is termed Point Spread Function as it clearly describes how the image of a single point can be spread into a diffused spot, when imaged through the microscope (Fig. 2a, b). In reciprocal space, the CTF shows how contrast oscillates between positive and negative values along all spatial frequencies. This oscillating behavior is directly visible when observing the power spectrum of a typical digitized electron micrograph, with a series of concentric characteristic annular shapes also termed Thon rings after the name of the physicist who first described them (Thon 1971). The reader interested in the transmission electron microscopy image formation model may read Philippsen et al. (2007a); Zhou et al. (1996).
-
Additive noise. Biological samples cannot be directly imaged in the microscope due to the high-vacuum environment. They need to be either embedded in some more resistant material as vitreous ice, plastic resins or in some heavy atom (negative stain). Besides, samples are often stabilized on a thin carbon or silicon nitride films that provide extra mechanical resistance. The amorphous structure of the film, and/or vitreous ice, plastic or staining produce a random structure superimposed on the projection image of the object being studied. This random structure is considered to be additive Gaussian noise (Sorzano et al. 2004b) and the overall Signal-to-Noise Ratio (SNR) can be as low as 1/3 (Fig. 2c).
-
Missing orientations. See Data collection geometry Section.

From top to bottom: a ideal projection of the bacteriorhodopsin structure at 1/3.5 Å−1 resolution. b Same projection affected by the CTF (assuming an accelerating voltage of 200 kV, 15,000 Å of underfocus, and a spherical aberration of 0.5 mm). c Same projection affected by CTF and noise (SNR = 1/3)
Sample preparation
Depending on the nature of samples, different techniques have been proposed for specimen preparation. Proteins or macromolecular complexes can be prepared from solutions using negative staining (Moritz et al. 2000), fast-freezing (Lepault and Dubochet 1986) or cryo-negative staining (Adrian et al. 1998). For cells, sectioning after chemical fixation and embedding in plastic resins is required. However, such preparation technique can damage the ultra-structure of biological samples (van Marle et al. 1995). Thus, new methods such as high-pressure freezing (Dubochet 1995) and cryo-fixation (Hsieh et al. 2002) provide better ultra-structural preservation of biological samples.
Negative staining is based on a highly scattering effect of heavy metal atoms on electrons which combined with small objective aperture can generate a high contrast. Hence, negative staining effect is obtained when a biological macromolecular complex is adsorbed on a carbon film of a sample grid in the presence of uranyl acetate, ammonium molybdate or phospho-tungstic acid, and let to dry out before being introduced in the microscope. With such preparation technique, an even layer of heavy atom salt covers the grid, except where biological complexes are adsorbed. This method is fast and produces images of biological macromolecules with a high contrast, depending on local stain thickness and stain exclusion by macromolecular complexes. However, samples are dehydrated and flattened, reducing the final quality and resolution of reconstructed volumes. Cryo-electron microscopy of frozen-hydrated specimens preserves natural hydratation of biological samples and provides some mechanical and chemical stability under the electron beam. To get vitreous ice, thin layers of sample solutions (∼100 nm) are rapidly frozen in liquid ethane (Dubochet et al. 1982). However, images of frozen-hydrated samples have a low contrast and reveal high specimen sensitivity to radiation. An intermediary approach reducing drawbacks of freezing in liquid ethane, is cryo-negative staining (Adrian et al. 1998). This technique combines negative staining with freezing in liquid ethane under hydrated conditions, resulting in an increased contrast, while preserving samples hydrated and slightly more protected against radiation damage (De Carlo et al. 2002). This method is potentially interesting for small macromolecular complexes hard to see in vitreous ice (less than 500 kDa). However, one must remember that because of the presence of stain, samples are not in physiological conditions, and interpretation of high-resolution contrast is still a subject of discussion.
For cellular samples, classic preparation methods require chemical fixation and embedding in plastic resins. However, as in the case of negative staining, this implies sample dehydratation generating artefacts. These artefacts can be reduced using physical fixation under high-pressure conditions (Hsieh et al. 2002; Matias et al. 2003) followed by plastic resin embedding or direct sectioning (Dubochet and Sartori Blanc 2001). The latter approach allows getting vitrified sections that can be observed in the microscope at liquid nitrogen temperature without any staining agent. This technique named CEMOVIS (Cryo-Electron Microscopy Of VItrified Sections) preserves the biological samples in near-native conditions (Al-Amoudi et al. 2004). However, the necessity of highly trained people on specific cryo-ultramicrotoms may be a limitation for a fast spreading of this method (Al-Amoudi et al. 2003).
Data acquisition
To select the appropriate technique for data acquisition, one should consider the nature of the specimen (Nature of the specimen Section), data collection geometry (Data collection geometry Section) and possibilities for automated data collection (Automated data collection Section).
Nature of the specimen
Depending on the nature of the specimen, a 3D structure reconstruction can be performed using one of the three following strategies:
-
Single object reconstruction. In this case, a single object is imaged in vitreous ice. This is a relatively new technique called Electron Tomography. The main advantage is that all projections (between 60 and 150) come from the same object, and therefore, they reflect the structure of a unique biological architecture. The drawback is that the object being imaged is severely damaged by the electron beam. The electron dose is kept as low as possible (the lower it is, the noisier the image is) to prevent structural alterations during the data acquisition.
-
Reconstruction of aperiodic objects. A possible solution to the radiation dose problem of the previous strategy is imaging many copies of structurally identical objects. In this approach, each object is imaged only once, and electron dose during exposure can be raised to 10 electrons Å−2 to get more contrasted images. The aperiodic nature of such samples comes from the random location and orientation of particles within the electron microscope field. An advantage of this technique is that we may have projection views from almost all directions if the orientation is sufficiently random. A disadvantage is that one has to estimate the direction of projection for each image because each particle orientation is random and unknown. The number of extracted particle images may vary from 1,000 to 100,000. This technique is commonly called single particle analysis.
-
Reconstruction of periodic objects. In this approach, multiple copies of the object under study are forced into a specific periodic spatial disposition. Thus, most uncertainties associated with particles random orientations can be avoided. Three most frequently observed arrangements of this sort are 2D crystals (different copies of the object are distributed over a 2D-plane), tubular assemblies with helical symmetry (different copies of the object are distributed over a cylinder), and a special case of single particle with icosahedral symmetry (naturally encountered in icosahedral viruses). The drawback of this latter approach is that it is not always possible to force this regular arrangement.
Data collection geometry
Ideally, we should acquire image projections of an object from all directions to determine its 3D structure (Kak and Slaney 1988). In practice, we have to relax this strong constraint of an infinite number of projections. The use of a finite set of projections and the high noise level make it difficult to recover the genuine 3D architecture of the sample. Therefore, one can only compute an approximate 3D structure, containing some artefacts and noise. If the object is unique by nature (as is the case in Electron Tomography), different views are acquired by tilting the specimen grid in the microscope around a fixed axis (this way of collecting data is called single-axis tilt geometry, Radermacher 1988). The more the specimen is tilted, the thicker it is. Hence, a practical tilting limit is reached around 70°. Above this value nearly all electrons are scattered and no projection image is produced.
The central-slice theorem relates the 2D Fourier transform of each projection image to the 3D Fourier transform of the object (Kak and Slaney 1988). This theorem states that “the Fourier transform of a 2D projection of any 3D object is equal to a central slice cutting through the origin of the 3D Fourier transform of this object, the slice being perpendicularly oriented with respect to the projection direction”. Therefore, as the maximum tilt of the specimen is limited, a region of Fourier space will always remain empty, inducing anisotropy in the 3D reconstruction volume. Such problem is called the missing wedge artefact. There are collection geometries that aim at reducing this missing wedge by using two (Mastronarde 1997) or multiple tilt axes (Messaoudi et al. 2006b).
In the case of multiple copies of differently oriented objects (as single particles, tubular assemblies with helical symmetry or icosahedral viruses), angular sampling is nearly unlimited since most projection directions can be generated without tilting the specimen grid.
Automated data collection
Electron microscopy of biological materials has recently experienced (and is still experiencing) a huge transformation involving the several steps of image acquisition. Automation of electron microscopes started in the early 1990s (Dierksen et al. 1992; Koster et al. 1992). Nowadays, robots that automatically load samples in the microscope have been developed (Potter et al. 2004). Microscopes can be automatically driven to find sample grids, select areas with proper vitreous ice thickness, autofocus and record images (Lei and Frank 2005; Suloway et al. 2005; Zhang et al. 2001). Specimen tilt can also be automatically monitored (Mastronarde 1997; Zheng et al. 2004; Ziese et al. 2003). In the same line, one can detect cryo-electron microscope images presenting defects (charging, drift, astigmatism) or simply a lack of detectable signal at high resolution (undetectable Thon rings even when using enhanced power spectrum algorithm) (Jonic et al. 2007). In single particle analysis, automatic procedures can select and extract particles from micrographs (for reviews on these algorithms see Nicholson and Glaeser 2001; Zhu et al. 2004). In tomographic data analysis, specific algorithms automatically align projection images and reconstruct 3D objects (Brandt et al. 2001a, b; Messaoudi et al. 2007). The combination of all these automatic procedures aim at computing a 3D map at sub-nanometer resolution within 24 h after inserting the specimen grid in the microscope (Zhu et al. 2001).
3D reconstruction
The reconstruction of an object from its 2D projections acquired in the electron microscope is usually performed in two steps: image alignment and tomographic reconstruction. Image alignment is specific to the nature of the data at hand and will be explained for each acquisition strategy (Single object reconstruction, Reconstruction from multiple copies of aperiodic objects, Reconstruction of periodic or highly symmetric objects Sections). Tomographic algorithms are described in Tomographic algorithms Section. All reconstruction algorithms make a number of assumptions that are reviewed in Reconstruction conditions Section.
Reconstruction conditions
Images collected for 3D reconstruction must meet three conditions. Failure to meet these conditions results in low resolution volumes and/or volumes with artefacts:
-
Homogeneity. Images must be 2D projections of the same object. This is an obvious statement that is not always easy to meet. As mentioned in Transmission electron microscopy principles Section, proteins may be flexible and, therefore, 2D projections of purified protein samples may contain complexes with slightly different conformations. Another obvious example is electron tomography (see below), where gradual sample degradation may occur during collection of tilt series.
-
Same numerical framework. Images are assumed to be projections of a single volume. Therefore, they must be “numerically compatible”. Hence, collected 2D projections must be normalized to compensate for different exposure times, different digitizers, or in the presence of local background gradients (Sorzano et al. 2004b).
-
Same geometrical framework. The projection direction corresponding to each image must be estimated as well as the relative in-plane shifts and rotations with respect to a centred volume. This process is usually known as image alignment and is specific to the nature of the object (see below). Single particle images are aligned using different algorithms than electron tomograms or crystal projections. These different alignment strategies will be explained in the following (Single object reconstruction, Reconstruction from multiple copies of aperiodic objects, and Reconstruction of periodic or highly symmetric objects Sections).
Tomographic algorithms
3D reconstruction can be accomplished in real space or in reciprocal space. Several families of reconstruction algorithms can be distinguished according to the reconstruction principle:
-
Fourier direct methods. Fourier reconstruction algorithms make use of the central-slice theorem to assemble Fourier transforms of 2D projections within an estimated 3D Fourier transform of the object. After inverse Fourier transformation, a 3D model of the object is obtained in real space (Kak and Slaney 1988). The computation of the 3D Fourier transform on a regular grid is a difficult interpolation problem.
-
Backprojection methods. Backprojection methods bring into play the central-slice theorem by rewriting the inverse Fourier transform in polar coordinates and rearranging integration limits (Kak and Slaney 1988). The weighted backprojection method (Radermacher 1992) comprises two steps: (1) multiplying Fourier transform of each 2D projection by a specific weighting function (the filtering part), and (2) summing the inverse Fourier transforms of weighted images (the backprojection part). These methods are usually more accurate than Fourier reconstruction methods since they perform interpolation in real space. Simple linear interpolation is usually satisfactory in the case of a backprojection algorithm while more complicated interpolation schemes are required for Fourier methods. Weighted backprojection methods have been specifically developed for conical-tilt geometry (Radermacher 1988, 1992). For single-axis tilt geometry, the weighted backprojection algorithm is equivalent to the standard filtered backprojection proportional to the frequency magnitude (Kak and Slaney 1988).
-
Series expansion methods. Series expansion methods express the reconstruction volume in real space as a linear combination of basis functions distributed on a regular spatial grid (Herman 1980). Expressing volumes in this way allows building a linear equation system whose unknowns are the basis weights and where there are as many equations as pixels available in the whole projection set. The two most popular series expansion methods in transmission electron microscopy are ART (Algebraic Reconstruction Technique) (Herman et al. 1973; Marabini et al. 1998) and SIRT (Simultaneous Iterative Reconstruction Technique) (Gilbert 1972; Penczek et al. 1992). Series expansion methods have been shown to be less affected by a low number of projections (Marabini et al. 1997, 2004) and by the presence of a missing region in Fourier space (Sorzano et al. 2001).
The reader interested may go through the following reviews on tomographic methods (Defrise 2001; Herman 1998).
Single object reconstruction
The reconstruction algorithms used in electron tomography make the assumption that projection images share a common line corresponding to the tilt axis. Therefore, the determination of this tilt axis becomes critical to obtain a correct reconstruction. In an ideal case there are no displacement of the object during images acquisition. Therefore, all points of the object move following parallel trajectories perpendicular to the tilt axis. The tilt axis can then be easily determined as a line that appears in the power spectrum of the sum of different projection images and its direction can be determined as the angle between this line and the vertical axis of the power spectrum. Before measuring the tilt axis, one may have to perform a translational alignment of projection images due to drifts occurring during acquisition. These drifts have a mechanical origin but may be coupled with charging effect or thermal drifts in case of samples observed at liquid nitrogen temperature.
-
A simple approach to minimize the drift effect during acquisition of different images belonging to a tilt series is based on cross-correlation. In this method, shifts between two consecutive images recorded at different angles are calculated, and the sample projection is centred before taking a new image. This procedure requires two acquisitions per angle, which increases the total electron dose received by the sample, and is mainly responsible for radiation damage in case of cryo-preparations.
-
A second approach consists in estimating the drift of consecutive images on a nearby area close to the zone of interest. As in the previous approach, the estimated drift is then corrected before recording the image at the current angle, but this method preserves the sample from severe radiation damage (Messaoudi et al. 2003).
-
A third approach consists in computing a model of the drift as a function of tilt for a given set of experimental conditions. Stored corrections of shift values are applied during data acquisition (Ziese et al. 2003).
Nevertheless, image alignment should be always checked before 3D reconstruction. Variations of at least ±5° in the tilt axis are commonly found when images are aligned after acquisition. The post-acquisition alignment can be performed by cross-correlation methods or by the use of recognizable features, such as gold particles, present in the sample. All public tomographic software (Kremer et al. 1996; Messaoudi et al. 2006a, 2007; Nickell et al. 2005) include methods to align images and determine the tilt axis geometry. Once the tilt series is aligned and the tilt parameters are determined, 3D reconstruction can be carried out, using any method described in Tomographic algorithms Section.
A major problem in electron tomography is the missing wedge phenomenon, corresponding to the lack of information imposed by the impossibility to record projections at high tilt angles (Hoppe and Hegerl 1980). The missing wedge can be minimized using specially designed high-tilt holders or using special acquisition geometries. The simplest of these geometries is the dual-axis approximation. In this approach, two perpendicular tilt-series of a single object are recorded and their corresponding 3D reconstructions combined (Mastronarde 1997; Penczek et al. 1995). An extension of this approach is the multiple-axis tomography. In this case, several tilt series are recorded after rotating the specimen in a set of in-plane selected angles (Messaoudi et al. 2006b). A third possible geometry is the conical tomography approach. In this case the tilt series are recorded by rotating the grid after tilting (Lanzavecchia et al. 2005; Zampighi et al. 2005). For general reviews of electron tomography the reader can check (Baumeister and Steven 2000; Koster and Klumperman 2003; Sali et al. 2003).
Example of reconstruction of basal bodies: Basal bodies are single organelles located at the base of cilia. These cilia are composed of an axoneme, which joins a basal body by a transition zone located at the membrane region in which the two central microtubules of the axoneme disappear, as has been demonstrated by recent tomographic studies performed on high-voltage electron microscopes (O’Toole et al. 2003). As single objects they cannot be analysed by single particle analysis and their size (250 nm in diameter and 450 nm in length cylinders) does not make possible their crystallization or analysis by RMN techniques. In addition, they present a structural complexity and dynamics that requires their study in situ inside cells. Therefore, electron tomography is the best method for their structural study.
Paramecia is a model organism for tomographic studies of basal bodies. Paramecium tetraurelia have a cortex covered with more than 4,000 motile cilia associated with their corresponding basal bodies. These basal bodies are regularly organized into parallel rows whose duplication is coordinated during cell cycle. Although the global structure of paramecia basal bodies has been the object of precise analysis by transmission electron microscopy (Dute and Kung 1978), their internal organization remains elusive. The most detailed internal structural information concerns the proximal end, called cartwheel. The cartwheel consists of a central hub and nine spokes which radiate to peripheral microtubules constituting the cylindrical structure. A tomographic study of Paramecium tetraurelia basal bodies, performed in multiple axis tomography provided new insights in this internal structure as well as in the organization of branching points of kinetodesmal fibres (Messaoudi et al. 2006b). These fibres are basal body appendages presumably involved in basal body anchoring and cohesion of the whole ciliature. They appear as three branching points located at defined microtubule triplets at the proximal end of basal bodies. Two of them are located at the same microtubule triplet but at different heights and the third one is placed at a contiguous microtubule triplet of the cylindrical structure of the basal body (Fig. 3). Regarding internal space of basal bodies, it presents the characteristic organization initially described by Dippell (1968, 1976) with several cartwheels in the proximal that may be identified by their central hub (Fig. 3), as well as a fine filamentous system associated with dense granules, which cover the distal 2/3 of the lumen, up to the terminal plate (Fig. 3), and which has been interpreted successively as nucleic acid (Dippell 1976) or glycogen (Mignot et al. 1993).

Central sections of tomographic reconstruction from Paramecia basal bodies. Planes has been extracted from a 310 × 310 × 200 voxels reconstruction each 5 from plane 60. In the central plane, cartwheels are boxed in green and internal dense material is boxed in blue. Kinetodesmal fibres are pointed by arrows. Scale bar 250 nm
For this example, tilt series from 300 nm epon sections of chemically fixed paramecia have been acquired in a Philips CM120 electron microscope, at a nominal magnification of 8,000×, and using an acceleration voltage of 120 kV. Six tilt series were recorded using a plug-in developed at our laboratory for automatic acquisition. This plug-in runs with Gatan Digital Micrograph (v3.1) software controlling a CCD Gatan camera (1 k, 24 μm). Each tomographic series (±45° with an angle increment of 1°) was acquired by rotating the grid in the horizontal plane at nominal angles of 0°, 30°, 45°, 60°, 90°, 120°. Images belonging to each tilt series were aligned and then divided into zones of size 512 × 512 pixels. The zones coming from a tilt series were subsequently aligned before computing six independent 3D reconstructions using weighted back-projection (see Tomographic algorithms Section). At the end, the volumes were combined using TomoJ software (Messaoudi et al. 2006a, 2007).
Reconstruction from multiple copies of aperiodic objects
The reconstruction from multiple copies of aperiodic objects is also known as single particle analysis. This approach assumes that the macromolecular complexes are trapped in all possible orientations, but in a single conformation. An advantage of this method is that a good angular coverage produces perfectly isotropic 3D reconstruction volumes. However, the angular coverage is sometimes reduced due to a preferential interaction of the molecule with the carbon film (Boisset et al. 1998). One disadvantage is that the projection direction must be estimated from the image itself. This step will be extensively discussed later. Using single particle analysis, 3D models with resolutions ranging between 1/15 and 1/6 Å−1 are routinely obtained for different specimens (e.g. icosahedral-symmetry viruses, D6 point group-symmetry haemoglobin, asymmetric Escherichia coli ribosome, etc.) (Falke et al. 2005; Fotin et al. 2006; Ludtke et al. 2004; Martin et al. 2007; Saban et al. 2006). The following reviews thoroughly analyse this topic Frank (2002, 2006), Schmid (2001), Subramaniam and Milne (2004), Tao and Zhang (2000), van Heel et al. (2000).
Usually a two-step procedure is used to compute a 3D particle model from windowed single-particle images:
-
1.
Image alignment. The position (the in-plane coordinates of the centre of the particle) and the orientation (one in-plane angle and two out-of-plane angles) are determined for each particle (Gelfand and Goncharov 1990; Goncharov 1990; Grigorieff 2007; Jonic et al. 2005; Penczek et al. 1994, 1996; Radermacher 1994; Radermacher et al. 1987; Sorzano et al. 2004c; van Heel 1987).
-
2.
3D reconstruction. A reconstruction algorithm computes a 3D model using centred particle images (shifted according to estimated translations) and calculated orientations. Algorithms from any of the families mentioned in Tomographic algorithms Section have been used in different single-particles approaches.
This workflow is iteratively executed until the model converges and angular assignment and resolution remain stable from one iteration to the next. Sometimes, the two steps are performed simultaneously (Provencher and Vogel 1988; Vogel and Provencher 1988; Yang et al. 2005), but the drawback of this approach is the large number of parameters to be estimated simultaneously, which conveys a higher risk of getting trapped into local optima of the objective function. There are two groups of techniques for determining the orientation and position of single particles:
-
1.
Reference free algorithms. This family of algorithms does not require any reference 3D model. The method of moments (Gelfand and Goncharov 1990; Goncharov 1990), the method of random conical tilt series (Radermacher et al. 1987), and the common-line technique (known also as angular reconstitution, or geometrical method) (Goncharov 1990; Penczek et al. 1994, 1996; van Heel 1987) belong to this group. The method of moments uses a known relationship between area moments of the 3D object and moments of its 2D projections. According to the method of random conical tilt series, pairs of images are recorded. Each pair consists of images recorded with tilted and untilted specimen. Extracted untilted-specimen images are subjected to multivariate statistical analysis and classification, while extracted tilted-specimen images (corresponding to the first exposure of the specimen to the beam) are used for reconstruction of a first 3D model. The common-line method requires the classification of projection images and computation of class averages to reduce noise. Geometrical relationships between class averages, in terms of two out-of-plane angles, are computed from the angles between 1D line projections that any two 2D projections of a 3D object have in common. The common-line projection for two 2D projections is found by comparing their sinograms line-by-line, and by identifying the maximum of the cross-sinogram correlation. At least, three different projections are required to orient images of an entirely asymmetric particle. This method has an equivalent in the Fourier space domain (Goncharov 1990).
-
2.
Algorithms needing a reference model. This group of algorithms refines particle orientation and position with respect to a reference volume using an iterative algorithm (Grigorieff 1998, 2007; Jonic et al. 2005; Penczek et al. 1994; Radermacher et al. 1994; Sorzano et al. 2004c). The operation of positioning an image plane with respect to a volume is known as image-to-volume alignment or 2D-to-3D registration. There are two types of reference-based techniques:
-
(a)
Discrete assignment. These methods determine the particle centre and orientation by a set of quantized parameters (Penczek et al. 1994; Radermacher et al. 1994; Sorzano et al. 2004c). These parameters are computed based on the cross-correlation between experimental images and a finite set of 2D projections of the reference volume, using their Fourier transforms (Penczek et al. 1994), wavelet transforms (Sorzano et al. 2004c) or Radon transforms (Radermacher et al. 1994). Their main drawback is that registration quality depends on the angular step used to compute projections of the reference volume (quantization step). The quantization step is reduced gradually during iterative refinement of the alignment parameters. The smaller the step, the more accurate, but slower is the parameter determination.
-
(b)
Continuous assignment. When close to the solution, one may avoid the dependence on a discrete library of projections, using continuous-parameter methods such as FREALIGN (Grigorieff 1998, 2007) or B-spline interpolation (Jonic et al. 2005). These methods are based on the central-slice theorem, and refine a set of initial values of particle centre and orientation in a space of continuous values by minimizing iteratively a measure of dissimilarity between the 2D Fourier transform of experimental images and extracted central slices of the 3D Fourier transform of the reference volume.
-
(a)
These and similar algorithms are incorporated in several image-processing packages used for single particle analysis, such as the commercial package IMAGIC (van Heel et al. 1996) and the open-source packages SPIDER (Frank et al. 1996), Xmipp (Sorzano et al. 2004a), EMAN (Ludtke et al. 1999), FREALIGN (Grigorieff 1998, 2007) and Bsoft (Heymann and Belnap 2007). The user can apply different reconstruction strategies within the same package; one may give better results than the others depending on the specimen.
Example of reconstruction of a multienzymatic complex: bacterial Glutamate synthase: We show here an example of single-particle reconstruction of glutamate synthase (GltS) at 1/9.5 Å−1 resolution. GltS is a multienzymatic complex composed of several copies of two types of subunits (alpha: 162 kDa and beta: 52.3 kDa). Present in bacteria and plants this complex is responsible for the main ammonia assimilation pathway. This example shows that the reconstruction strategy must be adapted to each sample. Also, it shows how a combined use of several complementary techniques can produce a 3D model at a sub-nanometer resolution. In this case, we used tilted- and untilted-specimen images by cryo-electron microscopy, single particle analysis, 3D reconstruction and CTF correction coupled with amplitude correction, using experimental small angle X-ray scattering (SAXS) data (Gabashvili et al. 2000; Svergun and Koch 2002).
First, as the stoichiometry of alpha and beta subunits and the global shape of the complex were unknown, we decided to use the random conical tilt series method to compute a first low resolution structure (Radermacher et al. 1987) (Fig. 4a). A set of twelve (45°/0°) tilted- and untilted-specimen images were collected on a Philips CM12 electron microscope, using an acceleration voltage of 120 kV, and defocus ranging between 1.5 to 1.7 μm). After digitization, image analysis and 3D reconstruction were mainly performed using the software package SPIDER and WEB (Frank et al. 1996). We used 437 pairs of interactively selected particles, boxed into 100 × 100 pixel images, centred and aligned. Moreover, untilted-specimen images were sorted in small homogeneous classes of well-defined electron microscope views (triangular top views, and rectangular side views) and their tilted-specimen counterparts were used to compute a first set of anisotropic 3D reconstruction volumes. To overcome the missing cone artefact, these volumes were aligned in real space in a common orientation and a merged reference volume was computed (Fig. 4a, Vref1). This volume was first refined with 2D–3D projection matching method (Penczek et al. 1994), and using 1,344 additional untilted-specimen images. At this stage, even if no symmetry were imposed during 3D reconstruction, it became clear that the complex assembly had a D3 point-group symmetry. Hence, a few additional refinement cycles were computed with imposed D3 symmetry, until resolution stabilized at a value of 1/26 Å−1 estimated with the Fourier Shell correlation (FSC0.5) criterion (Fig. 4a, Vref2). The volume obtained at this stage was perfectly isotropic and its overall shape was in a good agreement with a possible alpha:6 beta:6 stoichiometry for the GltS complex (Fig. 4b).

3D cryo-electron microscope single-particle reconstruction of a typical macromolecular complex: bacterial Glutamate synthase. a Single particle analysis strategy for the structural study. b Isosurface representation of side (left) and top (right) views of a 3D reconstruction at 1/26 Å−1 resolution. c Isosurface representation of side (left) and top (right) views of a 3D reconstruction at 1/9.5 Å−1 resolution. d Docking of atomic structures in the 3D reconstruction volume shown in c. The scale bar corresponds to 10 nm
To explore the architecture of the complex at higher (subnanometric) resolutions, additional set of cryoEM images were recorded on a JEOL JEM 2100F electron microscope equipped with a field emission gun. Images were recorded with a magnification of 50,000× an acceleration voltage of 200 kV, and with defocuses ranging from −1.7 to −3.2 μm. Images were recorded under low-dose conditions and digitized with a pixel size of 1.59Å × 1.59Å. We used enhanced diffractograms computed from experimental micrographs to semi-automatically detect and remove data introducing errors in the global 3D map (drifted micrographs, i.e. micrographs whose diffractograms contain diffraction rings truncated perpendicularly to the direction of the cryo-holder movement, caused by a thermal drift) or those unable to increase global SNR (micrographs whose diffractograms contain no diffraction rings) (Jonic et al. 2007). We rejected 68 of 152 collected micrographs, and selected 13,000 particles from the remaining 84 micrographs, using Roseman’s method (Roseman 2004). A high-resolution volume was obtained by projection matching, using the low resolution volume Vref2 as a starting reference structure. Hence, iterative projection matching was combined with CTF correction performed using Wiener filtering of volumes from focal series (Penczek et al. 1997). Defocus was computed for each image using the program CTFTILT (Mindell and Grigorieff 2003). The angular step (quantization) for projection matching was reduced gradually as the number of iterations was increasing. Also, different filters were applied on the reference volume at different stages of refinement, until resolution improved and stabilized at FSC0.5 = 1/9.5 Å−1.
We also had at our disposal experimental SAXS data of GltS computed by Dmitri Svergun (Svergun and Koch 2002). We corrected Fourier amplitudes of the reconstructed, non-filtered cryo-electron microscopy volume to fit those of SAXS data using the procedure described in (Gabashvili et al. 2000). Then, we filtered the corrected volume using a low-pass filter at 1/9.5 Å−1 (Fig. 4c). The atomic coordinates of the alpha subunit (PDB code: 1ea0, Binda et al. 2000) were then fitted in the volume, as well as a model of the beta subunit, derived from a homologous enzyme (PDB code: 1h7w, Dobritzsch et al. 2001) (Fig. 4d). A manuscript presenting a detailed description of this project and the docking of atomic structures is in preparation by Cottevieille et al.
Reconstruction of periodic or highly symmetric objects
If we have multiple copies of an object in a cryo-electron microscope field and if they adopt a regular spatial distribution, we can exploit this knowledge in order to improve particle alignment. This is the case of biological 2D crystals, helical particles, and icosahedral virus shells. In all these cases, the protein or sets of proteins being studied are regularly ordered: in a plane (2D crystals), in a cylinder (helical particles), or in an icosahedron (icosahedral viruses). Alignment and reconstruction techniques used in each case are different. Currently, the use of regular arrangement is the only possibility of achieving nearly atomic resolution using electron microscopy (Gonen et al. 2005).
Single-layer biological 2D crystals: Electron Crystallography
Many membrane proteins can naturally produce 2D crystals (Walz and Grigorieff 1998) or can be artificially forced to crystallize in specific conditions (Berry et al. 2006). The study of biological 2D crystals goes back to the early 1970s (Matricardi et al. 1972; Taylor and Glaeser 1974; Unwin and Henderson 1975). Recent studies achieve resolutions ranging between 1/5.5 and 1/1.9 Å−1 (Gonen et al. 2005; Mitsuoka et al. 1999; Ruprecht et al. 2004).
The experiment takes advantage of the electron diffraction. The crystal quality (periodicity, structural homogeneity, etc.) is directly related to the quality of the diffraction information. The diffraction pattern is recorded in real space and usually processed in reciprocal space using the Fourier transform of the recorded image. Because of the periodicity of the structure, the diffraction image contains a set of peaks (called spots) distributed on a regular lattice called the reciprocal lattice (to be distinguished from the crystal lattice defining the regular disposition of sample molecules in real space). As several crystals are generally observed at a fixed tilt angle, there is a missing cone artefact responsible for anisotropic resolution of the global 3D reconstruction volume. To counteract this missing cone artefact, additional information (e.g. any a priori knowledge) must be taken into account during 3D reconstruction. One of the most used methods is solvent flattening (Wang 1985), which assumes that the solvent outside the protein has a constant intensity value. The image-processing workflow for Electron Crystallography comprises the following steps:
-
1.
Noise filtering. This step is usually performed by masking out all those values in Fourier space far from a reciprocal lattice spot. This step is usually called optical filtering.
-
2.
Correction of crystal defects. Biological crystals are far from ideal mathematical crystals. Usually they exhibit local crystal patches (i.e. the crystal is not formed by a unique and uniform crystal but by several crystal patches with different orientations) intermixed with amorphous regions. The aim of this step is to find genuine crystal areas and to re-interpolate the recorded image so that the whole area can be considered as a perfect crystal (Gil et al. 2006).
-
3.
Projection alignment. Assuming that the projection direction of each image is known, this step aims at determining the relative shifts of different images. This step is usually known as phase origin determination since a shift in real space causes a phase shift in Fourier space.
-
4.
Direct Fourier reconstruction. The most common reconstruction algorithm in Electron Crystallography is Fourier inversion of the 3D Fourier transform. The latter is interpolated from the diffraction spots available in projections. Given the single-axis tilt geometry, it can be easily proved that different spots in all images lie along certain lines. For this reason the interpolation is carried out along these lines for which a simple 1D interpolation problem is solved.
The software packages most commonly used for 2D crystal reconstruction are the MRC Image Processing Package (Crowther et al. 1996), 2dx (Gipson et al. 2007), IPLT (Philippsen et al. 2007b), Bsoft (Heymann and Belnap 2007) and Xmipp (Sorzano et al. 2004a). The reader interested in this topic may also study the reviews (Ellis and Hebert 2001; Fernandez et al. 2006; Glaeser 1999; Walz and Grigorieff 1998).
Helical particles
Some proteins tend to form helical assemblies (i.e. flagella or filaments). Helical crystals can be thought of as 2D crystals that have been rolled on a cylinder, and for this reason, they share many commonalities with 2D crystal image processing. The first structure of this kind reconstructed by electron diffraction was studied in 1968 (DeRosier and Klug 1968) although the X-ray diffraction of helices was known earlier (Klug et al. 1958). The algorithmic workflow was quickly established in 1970 (DeRosier and Moore 1970) and later revised (Morgan and Rosier 1992). However, this field is rather stable and new developments are not as active as in other electron microscope fields. Helical crystals are naturally disordered because they tend to flex in solution (Egelman 1986), different parts of the helix may have different pitches (angular disorder) (Wang et al. 2006), and because of specimen flattening on the support film (Morgan and Rosier 1992). These disorders make helical crystals weak diffractors. For many years, the resolution achieved with this kind of data was medium-to-low (from 1/25 to 1/9 Å−1). However, more recent image-processing developments as well as improvements in sample preparation techniques produced nearly atomic resolutions (1/4 Å−1, Miyazawa et al. 2003).
A helical object can be expressed as a sum of sinusoidal helical waves (Fourier–Bessel decomposition) (Klug et al. 1958). In the Fourier space, the Fourier transform of an object is zero everywhere except at given planes (called layer planes). The central-slice theorem states that any projection perpendicular to the helical axis is a central slice of the 3D Fourier transform of the helical object that contains the helical axis in reciprocal space. Therefore, the 2D Fourier transform of the projection is zero everywhere except at some lines (the intersection of the central slice with the layer planes) called layer lines. Moreover, knowing the value of the Fourier transform at a point of the layer line allows the computation of all values belonging to the layer plane and at the same radial distance from the reciprocal helical axis (i.e. of all values within the circumference to which the point in the layer line belongs) (Wang and Nogales 2005).
The workflow used in helical reconstruction is similar to that used in 2D crystal reconstruction:
-
1.
Noise filtering. This step is usually performed by masking out all values in the Fourier space that do not lie within layer lines. This step is the equivalent to the optical filtering in 2D crystals.
-
2.
Correction of non helical features. This step is equivalent to crystal unbending for 2D crystals. It provides algorithms for correction of departures from a straight helical symmetry (Egelman 1986) and corrections of non constant pitch (Bluemke et al. 1988). To our knowledge, sample flattening is not explicitly corrected. However, it is carefully monitored (Morgan and Rosier 1992) in order to keep artefacts within reason.
-
3.
Determination of the particle axis and correction of the out-of-plane tilt. In this step, the particle helical axis is determined. The out-of-plane tilt is estimated from the image and Fourier coefficients are corrected for this effect.
-
4.
Projection alignment. Relative shifts among different projections must be found (as in the 2D case). In the reconstruction of helical structures, it is common to use several projection images of similar structures. The relative tilt between structures must also be determined. Once all projections are aligned (shift and tilt), the corresponding layer lines are averaged to produce a cleaner estimate of the Fourier coefficients on those lines.
-
5.
Direct Fourier reconstruction. As in the 2D crystal case, the most widespread reconstruction algorithm is Fourier inversion. This is done by interpolating Fourier coefficients in the 3D frequency space.
The most widely used software packages for processing helical structures are the MRC Image Processing Package (Crowther et al. 1996), the Brandeis Helical package (Owen et al. 1996) and PHOELIX (Carragher et al. 1996). For further reading, see DeRosier and Moore (1970), Egelman (2007), Henderson (2004), Morgan and Rosier (1992), Wang and Nogales (2005).
Icosahedral particles
The icosahedral symmetry of some virus capsids may be considered a special case of periodic objects (Baker et al. 1999; Grunewald and Cyrklaff 2006; Lee and Johnson 2003; Thuman-Commike and Chiu 2000). As in the previous periodic cases (2D crystals and helical particles), this regular arrangement of proteins can be used in order to increase the SNR of the reconstructed volume. It should be noted that only the capsid follows icosahedral symmetry while the virus inner parts and/or virus tails do not. However, all this internal and external non-symmetric structures are lost when using algorithms for symmetric reconstruction. Resolutions as high as 1/6.5 Å−1 can be achieved with this technique (Zhou et al. 2001), although most studies remain in the range between 1/25 and 1/10 Å−1 (Lee and Johnson 2003). Multiple copies of the same virus are imaged and combined for 3D reconstruction. The most common image-processing workflow for icosahedral particles is:
-
1.
Obtention of an initial low resolution model. An initial centre and orientation determination is done for a small subset of images (between 5 and 10, Thuman-Commike and Chiu 2000). Relying on icosahedral symmetry, each projection of the virus capsid provides other 59 equivalent projections (with different projection directions). This redundancy dramatically increases the amount of information available multiplying by 60 the number of original images. This means that any pair of images shares a maximum of 60 common lines (they may share less, depending on whether they fall or not nearby symmetry axes). For this reason, image alignment by common lines is one of the most used algorithms (Crowther 1971; Crowther et al. 1970; Fuller et al. 1996; Thuman-Commike and Chiu 1997). Once an initial guess for the alignment parameters is obtained from a small image set, a low-resolution 3D reconstruction is computed and used as a reference for more refined structures.
-
2.
Refinement of the reconstructed model. A single particle analysis alignment approach is followed with hundreds or thousands projections in order to increase the resolution. The orientation and shift of each projection is compared to projections of the reference model within the asymmetric unit. The new alignment parameters are used for reconstructing a new model. This process is iterated until stable convergence is reached. The 3D reconstruction can be performed by direct Fourier inversion (Crowther et al. 1970) or using any real-space methods (Scheres et al. 2005a). As in the case of single particle analysis, virus capsids are also strongly affected by intrinsic variability since this is a part of the virus life cycle (Aramayo et al. 2005). For this reason, the analysis of variability in viruses is currently an important issue (Scheres et al. 2005a). Some efforts are recently taken in the direction of performing non-symmetric reconstructions of viruses (even if they are icosahedral) (Grunewald et al. 2003). In this way, a non-symmetric structure (interior and exterior tails) can also be reconstructed. Even Electron Tomography approaches (Grunewald et al. 2003) are being used to visualize the virus cycle within infected cells.
The most used software packages for reconstructing icosahedral viruses are the MRC Image Processing Package (Crowther et al. 1996), IMAGIC (van Heel et al. 1996), EMPFT (Baker and Cheng 1996) and SPIDER (Frank et al. 1996). For further reading on this topics, see (Baker et al. 1999; Conway and Steven 1999; Navaza 2003; Thuman-Commike and Chiu 2000).
CTF correction
Techniques for computational CTF correction require accurate estimation of parameters such as defocus, astigmatism, defocus spread and envelope. Many methods have been developed and they all require an estimation of the power spectrum, obtained either by classical methods such as periodogram averaging (Fernandez et al. 1997) or by parametric methods such as AR or ARMA (Fernandez et al. 1997; Velazquez-Muriel et al. 2003). In practice, periodogram averaging is simple and fast, although estimates may be quite noisy (Broersen 2000). On the other hand, parametric methods are more complex and slower although the power spectrum estimate may be more accurate (Broersen 2000; Velazquez-Muriel et al. 2003).
Once a power spectrum estimate is computed, CTF parameters are usually estimated by minimizing a measure of dissimilarity between experimental and theoretical power spectra. Some works concentrate on the estimation of defocus parameters, astigmatism, and contrast amplitude factor (Mindell and Grigorieff 2003; Toyoshima and Unwin 1988; Toyoshima et al. 1993). Other works estimate amplitude decay either using a Gaussian envelope (specified by a parameter called B-factor) (Huang et al. 2003; Mallick et al. 2005; Saad et al. 2001; Sander et al. 2003) or by fitting parameters of a physical model (Velazquez-Muriel et al. 2003; Zhou et al. 1996; Zhu et al. 1997). Some of these works minimize the dissimilarity between modeled and experimental power spectra, previously circularly averaged (Saad et al. 2001; Zhou et al. 1996) or elliptically averaged (Mallick et al. 2005), while other works propose a full 2D optimization (Mindell and Grigorieff 2003; Velazquez-Muriel et al. 2003). The work of Huang et al. lies somewhere in between a full 2D optimization and a 1D optimization since astigmatism is estimated in averaged sectors (Huang et al. 2003).
Once CTF parameters are accurately estimated, the CTF can be corrected. The simplest method for CTF correction is phase flipping. This is carried out by shifting phases at 180° for frequencies where contrast is negative (Frank 2006). Both amplitude and phase can be corrected with the following methods: Wiener filtering of images (Frank and Penczek 1995; Grigorieff 1998), Wiener filtering of volumes computed from focal series (Penczek et al. 1997), Iterative data refinement (Sorzano et al. 2004d), Maximum entropy (Skoglund et al. 1996), Direct deconvolution in Fourier space (Stark et al. 1997), Chahine’s method (Zubelli et al. 2003), etc.
Current developments and prospect
Image analysis
From the image-analysis point of view, there are several topics that deserve further investigation in order to make electron microscopy a mature technique with a fully productive capacity:
-
Fully automated process. Although some works have already been reported about automation of the 3D reconstruction process (starting from the sample preparation and loading into the microscope to the final 3D reconstruction) (Zhu et al. 2001), the field is still far from a high throughput scenario. Many steps (particularly, automatic particle picking and classification) must still be more robust and reliable.
-
Software interoperability. As has already been shown, many software packages can be used for performing each step of the image processing workflow. Each package has its own strengths and weaknesses. An ideal workflow should make use of the strongest points of each package. However, moving data from one package to another is rather troublesome, not only because of different file formats but also because of different internal spatial conventions. Some efforts have recently been made to facilitate data interchange (Heymann et al. 2005). However, the ideal of a transparent workflow design using programs from several software packages is still far from reality.
-
Reconstruction post-processing. Currently, the 3D reconstruction is the final product of image processing analysis. However, in some situations, one may require a boost of the information conveyed by this volume. For example, to improve the biological understanding of these structures, one should further address the following compulsory steps: volume deconvolution (to get rid not only of the CTF but also of algorithm and collection geometry artifacts), volume denoising and volume segmentation.
-
Database depositions. The current existence of structural databases such as the European Macromolecular Structure Database (Boutselakis et al. 2003) and the requirement from some journals to deposit resolved structures before publication will force software packages to provide means to facilitate the submission of structures and of workflow parameters.
Relationship with other structural data sources
The routine combination of electron microscopy data with other structural resources is another topic that will be presumably very active in the near future. Fitting of high resolution structures into low/medium resolution electron microscope maps is becoming more and more popular (Tama et al. 2004a, b; Velazquez-Muriel and Carazo 2007; Volkmann and Hanein 1999). The fitting may be rigid or it may take into account the flexibility of proteins. This approach is also complemented with the prediction of structural folding (Baker and Sali 2001; Jones 2001) or folding super families (Velazquez-Muriel et al. 2005). However, fitting high resolution structures into low/medium resolution volumes is still quite human-dependent and fully automatic and reliable methods are still under development.
The information provided by cryo-electron microscopy may be complemented by other information sources such as Atomic Force Microscopy (Dimmeler et al. 2001; Engel and Muller 2000) or SAXS (Hamada et al. 2007) that help to constrain the search space in the reconstruction process. This extra information should result in an improvement of resolution, as long as the two sources of information correspond to the same conformation of the particle (Vestergaard et al. 2005).
An extension of electron tomography is its combination with chemical mapping by energy-loss imaging (Leapman et al. 2004; Mobus et al. 2003) that will have important applications in material sciences and in biology in a near future. This allows the spatial localization of chemical components. For this purpose, acquisition of tilt series from energy filtered electrons is required. The main limitation of this method is the sensitivity of biological samples to beam damage. Moreover, calculation of characteristic signal requires a background subtraction and the registration of tilt series acquired at different energy loss values (Boudier et al. 2005).
Finally, the information provided by electron microscopy should be integrated in larger structures. As is already the case of molecular structures fitted in electron tomograms (Böhm et al. 2000; Frangakis et al. 2002; Nickell et al. 2006), electron tomograms should be integrated in even larger volumes obtained by X-ray tomography (Le Gros et al. 2005; Meyer-Ilse et al. 2001) or even optical microscopy. This combination of information is giving raise to new fields such as correlative microscopy (Leapman 2004) and visual proteomics (Nickell et al. 2006).
References
Adrian M, Dubochet J, Fuller SD, Harris JR (1998) Cryo-negative staining. Micron 29:145–160
Al-Amoudi A, Chang JJ, Leforestier A, McDowall A, Salamin LM, Norlen LP, Richter K, Blanc NS, Studer D, Dubochet J (2004) Cryo-electron microscopy of vitreous sections. Embo J 23:3583–3588
Al-Amoudi A, Dubochet J, Gnaegi H, Luthi W, Studer D (2003) An oscillating cryo-knife reduces cutting-induced deformation of vitreous ultrathin sections. J Microsc 212:26–33
Aramayo R, Merigoux C, Larquet E, Bron P, Perez J, Dumas C, Vachette P, Boisset N (2005) Divalent ion-dependent swelling of tomato bushy stunt virus: a multi-approach study. Biochim Biophys Acta 1724:345–354
Baker D, Sali A (2001) Protein structure prediction and structural genomics. Science 294:93–96
Baker TS, Cheng RH (1996) A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol 116:120–130
Baker TS, Olson NH, Fuller SD (1999) Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol 63:862–922
Baumeister W, Steven AC (2000) Macromolecular electron microscopy in the era of structural genomics. Trends Biochem Sci 25:624–631
Berry IM, Dym O, Esnouf RM, Harlos K, Meged R, Perrakis A, Sussman JL, Walter TS, Wilson J, Messerschmidt A (2006) SPINE high-throughput crystallization, crystal imaging and recognition techniques: current state, performance analysis, new technologies and future aspects. Acta Crystallogr D Biol Crystallogr 62:1137–1149
Binda C, Bossi RT, Wakatsuki S, Arzt S, Coda A, Curti B, Vanoni MA, Mattevi A (2000) Cross-talk and ammonia channeling between active centers in the unexpected domain arrangement of glutamate synthase. Structure 8:1299–1308
Bluemke DA, Carragher B, Josephs R (1988) The reconstruction of helical particles with variable pitch. Ultramicroscopy 26:255–270
Böhm J, Frangakis AS, Hegerl R, Nickell S, Typke D, Baumeister W (2000) Toward detecting and identifying macromolecules in a cellular context: template matching applied to electron tomograms. Proc Natl Acad Sci USA 97:14245–14250
Boisset N, Penczek P, Taveau JC, You V, Haas Fd, Lamy J (1998) Overabundant single-particle electron microscope views induce a three-dimensional reconstruction artifact. Ultramicroscopy 74:201–207
Boudier T, Lechaire JP, Frebourg G, Messaoudi C, Mory C, Colliex C, Gaill F, Marco S (2005) A public software for energy filtering transmission electron tomography (EFTET-J): application to the study of granular inclusions in bacteria from Riftia pachyptila. J Struct Biol 151:151–159
Boutselakis H, Dimitropoulos D, Fillon J, Golovin A, Henrick K, Hussain A, Ionides J, John M, Keller PA, Krissinel E, McNeil P, Naim A, Newman R, Oldfield T, Pineda J, Rachedi A, Copeland J, Sitnov A, Sobhany S, Suarez-Uruena A, Swaminathan J, Tagari M, Tate J, Tromm S, Velankar S, Vranken W (2003) E-MSD: the European bioinformatics institute macromolecular structure database. Nucleic Acids Res 31:458–462
Brandt S, Heikkonen J, Engehardt P (2001a) Automatic alignment of transmission electron microscope tilt series without fiducial markers. J Struct Biol 136:201–213
Brandt S, Heikkonen J, Engehardt P (2001b) Multiphase method for automatic alignment of transmission electron microscope images using markers. J. Struct Biol 133:10–22
Brink J, Ludtke SJ, Kong Y, Wakil SJ, Ma J, Chiu W (2004) Experimental verification of conformational variation of human fatty acid synthase as predicted by normal mode analysis. Structure 12:185–191
Broersen PMT (2000) Facts and fiction in spectral analysis. IEEE Trans Instrum Meas 49:766–772
Carragher B, Whittaker M, Milligan RA (1996) Helical processing using PHOELIX. J Struct Biol 116:107–112
Conway JF, Steven AC (1999) Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J Struct Biol 128:106–118
Crowther RA (1971) Procedures for three-dimensional reconstruction of spherical viruses by Fourier synthesis from electron micrographs. Philos Trans R Soc Lond B Biol Sci 261:221–230
Crowther RA, Amos LA, Finch JT, Rosier DJD, Klug A (1970) Three dimensional reconstructions of spherical viruses by fourier synthesis from electron micrographs. Nature 226:421–425
Crowther RA, Henderson R, Smith JM (1996) MRC image processing programs. J Struct Biol 116:9–16
De Carlo S, El-Bez C, Alvarez-Rua C, Borge J, Dubochet J (2002) Cryo-negative staining reduces electron-beam sensitivity of vitrified biological particles. J Struct Biol 138:216–226
Defrise M (2001) A short reader’s guide to 3D tomographic reconstruction. Comput Med Imaging Graph 25:113–116
DeRosier D, Klug A (1968) Reconstruction of three-dimensional structures from electron micrographs. Nature 217:130–134
DeRosier DJ, Moore PB (1970) Reconstruction of three-dimensional images from electron micrographs of structures with helical symmetry. J Mol Biol 52:355–369
Dierksen K, Typke D, Hegerl R, Koster AJ, Baumeister W (1992) Towards automatic electron tomography. Ultramicroscopy 40:71–87
Dimmeler E, Marabini R, Tittmann P, Gross H (2001) Correlation of topographic surface and volume data from 3D electron microscopy. J Struct Biol 136:20–29
Dippell R (1968) The development of basal bodies in paramecium. Proc Natl Acad Sci USA 61:461–468
Dippell R (1976) Effect of nuclease and protease digestion on the ultrastructure of Paramecium basal bodies. J Cell Biol 69:622–637
Dobritzsch D, Schneider G, Schnackerz KD, Lindqvist Y (2001) Crystal structure of dihydropyrimidine dehydrogenase, a major determinant of the pharmacokinetics of the anti-cancer drug 5-fluorouracil. Embo J 20:650–660
Dubochet J (1995) High-pressure freezing for cryoelectron microscopy. Trends Cell Biol 5:366–368
Dubochet J, Lepault J, Freeman R, Berriman JA, Homo JC (1982) Electron microscopy of frozen water and aqueous solutions. J Microsc 128:219–237
Dubochet J, Sartori Blanc N (2001) The cell in absence of aggregation artifacts. Micron 32:91–99
Dute R, Kung C (1978) Ultrastructure of the proximal region of somatic cilia in Paramecium tetraurelia. J Cell Biol 78:451–464
Egelman EH (1986) An algorithm for straightening images of curved filamentous structures. Ultramicroscopy 19:367–373
Egelman EH (2007) The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol 157:83–94
Egerton RF, Li P, Malac M (2004) Radiation damage in the TEM and SEM. Micron 35:399–409
Ellis MJ, Hebert H (2001) Structure analysis of soluble proteins using electron crystallography. Micron 32:541–550
Engel A, Muller DJ (2000) Observing single biomolecules at work with the atomic force microscope. Nat Struct Biol 7:715–718
Falke S, Tama F, Brooks CL 3rd, Gogol EP, Fisher MT (2005) The 13 Å structure of a chaperonin GroEL-protein substrate complex by cryo-electron microscopy. J Mol Biol 348:219–230
Fernandez JJ, Sanjurjo JR, Carazo JM (1997) A spectral estimation approach to contrast transfer function detection in electron microscopy. Ultramicroscopy 68:267–295
Fernandez JJ, Sorzano COS, Marabini R, Carazo JM (2006) Image processing and 3-D reconstruction in electron microscopy. IEEE Signal Process Mag 23:84–94
Fotin A, Kirchhausen T, Grigorieff N, Harrison SC, Walz T, Cheng Y (2006) Structure determination of clathrin coats to subnanometer resolution by single particle cryo-electron microscopy. J Struct Biol 156:453–460
Frangakis AS, Bohm J, Forster F, Nickell S, Nicastro D, Typke D, Hegerl R, Baumeister W (2002) Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc Natl Acad Sci USA 99:14153–14158
Frank J (2002) Single-particle imaging of macromolecules by cryo-electron microscopy. Annu Rev Biophys Biomol Struct 31:303–319
Frank J (2006) Three-dimensional electron microscopy of macromolecular assemblies: visualization of biological molecules in their native state. Oxford University Press, New York
Frank J, Penczek PA (1995) On the correction of the contrast transfer function in biological electron microscopy. Optik 98:125–129
Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 116:190–199
Frank J, Verschoor A, Boublik M (1981) Computer averaging of electron micrographs of 40S ribosomal subunits. Science 214:1353–1355
Fuller SD, Butcher SJ, Cheng RH, Baker TS (1996) Three-dimensional reconstruction of icosahedral particles–the uncommon line. J Struct Biol 116:48–55
Gabashvili IS, Agrawal RK, Spahn CM, Grassucci RA, Svergun DI, Frank J, Penczek P (2000) Solution structure of the E. coli 70S ribosome at 11.5 Å resolution. Cell 100:537–549
Gao H, Valle M, Ehrenberg M, Frank J (2004) Dynamics of EF-G interaction with the ribosome explored by classification of a heterogeneous cryo-EM dataset. J Struct Biol 147:283–290
Gelfand MS, Goncharov AB (1990) Spatial rotational alignment of identical particles given their projections: theory and practice. Transl Math Monogr 81:97–122
Gil D, Carazo JM, Marabini R (2006) On the nature of 2D crystal unbending. J Struct Biol 156:546–555
Gilbert P (1972) Iterative methods for the three-dimensional reconstruction of an object from projections. J Theor Biol 36:105–117
Gipson B, Zeng X, Zhang ZY, Stahlberg H (2007) 2dx—user-friendly image processing for 2D crystals. J Struct Biol 157:64–72
Glaeser RM (1971) Limitations to significant information in biological electron microscopy as a result of radiation damage. J Ultrastruct Res 36:466–482
Glaeser RM (1999) Review: electron crystallography: present excitement, a nod to the past, anticipating the future. J Struct Biol 128:3–14
Goncharov AB (1990) Three-dimensional reconstruction of arbitrarily arranged identical particles given their projections. Transl Math Monogr 81:67–95
Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T (2005) Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature 438:633–638
Grigorieff N (1998) Three-dimensional structure of bovine NADH: ubiquinone oxidoreductase (complex I) at 22 A in ice. J Mol Biol 277:1033–1046
Grigorieff N (2007) FREALIGN: high-resolution refinement of single particle structures. J Struct Biol 157:117–125
Grunewald K, Cyrklaff M (2006) Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr Opin Microbiol 9:437–442
Grunewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC (2003) Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302:1396–1398
Gue M, Marco S, Croissy A, Rigaud JL, Boudier T (2005) Integrative imaging: a new challenge for cell biology. Imaging Microsc 7:51–53
Hamada D, Higurashi T, Mayanagi K, Miyata T, Fukui T, Iida T, Honda T, Yanagihara I (2007) Tetrameric structure of thermostable direct hemolysin from vibrio parahaemolyticus revealed by ultracentrifugation, small-angle X-ray scattering and electron microscopy. J Mol Biol 365:187–195
Harauz G, van Heel M (1986) Exact filters for general geometry three dimensional reconstruction. Optik 73:146–156
Henderson R (2004) Realizing the potential of electron cryo-microscopy. Q Rev Biophys 37:3–13
Herman GT (1980) Image reconstruction from projections: the fundamentals of computerized tomography. Academic, New York
Herman GT (1998) Algebraic reconstruction techniques in medical imaging. In: Leondes CT (ed) Medical imaging, systems techniques and applications: computational techniques, vol 6. Gordon and Breach Science Publishers, Amsterdam, pp 1–42
Herman GT, Lent A, Rowland SW (1973) ART: mathematics and applications. A report on the mathematical foundations and on the applicability to real data of the algebraic reconstruction techniques. J Theor Biol 42:1–32
Heymann JB, Belnap DM (2007) Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18
Heymann JB, Chagoyen M, Belnap DM (2005) Common conventions for interchange and archiving of three-dimensional electron microscopy information in structural biology. J Struct Biol 151:196–207
Hoppe W, Hegerl R (1980) Three-dimensional structure determination by electron microscopy (nonperiodic specimens). In: Hawkes PW (ed) Computer processing of electron microscope images, vol 13. Springer-Verlag, Berlin, pp 127–183
Hsieh CE, Marko M, Frank J, Mannella CA (2002) Electron tomographic analysis of frozen-hydrated tissue sections. J Struct Biol 138:63–73
Huang Z, Baldwin PR, Mullapudi S, Penczek PA (2003) Automated determination of parameters describing power spectra of micrograph images in electron microscopy. J Struct Biol 144:79–94
Jones DT (2001) Protein structure prediction in genomics. Brief Bioinform 2:111–125
Jonic S, Sorzano CO, Cottevieille M, Larquet E, Boisset N (2007) A novel method for improvement of visualization of power spectra for sorting cryo-electron micrographs and their local areas. J Struct Biol 157:156–167
Jonic S, Sorzano CO, Thevenaz P, El-Bez C, De Carlo S, Unser M (2005) Spline-based image-to-volume registration for three-dimensional electron microscopy. Ultramicroscopy 103:303–317
Kak AC, Slaney M (1988) Principles of computerized tomographic imaging. IEEE Press, New York
Klug A, Crick FHC, Wyckoff HW (1958) Diffraction by helical structures. Acta Crystallogr 11:199–213
Koster AJ, Chen H, Sedat JW, Agard D (1992) Automated microscopy for electron tomography. Ultramicroscopy 46:207–227
Koster AJ, Klumperman J (2003) Electron microscopy in cell biology: integrating structure and function. Nat Cell Biol Suppl S:SS6–SS10
Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76
Lanzavecchia S, Cantele F, Bellon PL, Zampighi L, Kreman M, Wright E, Zampighi GA (2005) Conical tomography of freeze-fracture replicas: a method for the study of integral membrane proteins inserted in phospholipid bilayers. J Struct Biol 149:87–98
Le Gros MA, McDermott G, Larabell CA (2005) X-ray tomography of whole cells. Curr Opin Struct Biol 15:593–600
Leapman RD (2004) Novel techniques in electron microscopy. Curr Opin Neurobiol 14:591–598
Leapman RD, Kocsis E, Zhang G, Talbot TL, Laquerriere P (2004) Three-dimensional distributions of elements in biological samples by energy-filtered electron tomography. Ultramicroscopy 100:115–125
Lee KK, Johnson JE (2003) Complementary approaches to structure determination of icosahedral viruses. Curr Opin Struct Biol 13:558–569
Lei J, Frank J (2005) Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J Struct Biol 150:69–80
Lepault J, Dubochet J (1986) Electron microscopy of frozen hydrated specimens: preparation and characteristics. Methods Enzymol 127:719–730
Leschziner AE, Nogales E (2006) The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol 153:284–299
Leschziner AE, Nogales E (2007) Visualizing flexibility at molecular resolution: analysis of heterogeneity in single-particle electron microscopy reconstructions. Ann Rev Biophys Biomol Struct 36:43–62
Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128:82–97
Ludtke SJ, Chen DH, Song JL, Chuang DT, Chiu W (2004) Seeing GroEL at 6 Å resolution by single particle electron cryomicroscopy. Structure 12:1129–1136
Mallick SP, Carragher B, Potter CS, Kriegman DJ (2005) ACE: automated CTF estimation. Ultramicroscopy 104:8–29
Marabini R, Herman GT, Carazo JM (1998) 3D reconstruction in electron microscopy using ART with smooth spherically symmetric volume elements (blobs). Ultramicroscopy 72:53–65
Marabini R, Rietzel E, Schröder R, Herman GT, Carazo JM (1997) Three-dimensional reconstruction from reduced sets of very noisy images acquired following a single-axis tilt schema: application of a new three-dimensional reconstruction algorithm and objective comparison with weighted backprojection. J Struct Biol 120:363–371
Marabini R, Sorzano COS, Matej S, Fernandez JJ, Carazo JM, Herman GT (2004) 3D reconstruction of 2D crystals. IEEE Trans Image Process 13:549–561
Martin AG, Depoix F, Stohr M, Meissner U, Hagner-Holler S, Hammouti K, Burmester T, Heyd J, Wriggers W, Markl J (2007) Limulus polyphemus hemocyanin: 10 A cryo-EM structure, sequence analysis, molecular modelling and rigid-body fitting reveal the interfaces between the eight hexamers. J Mol Biol 366:1332–1350
Mastronarde DN (1997) Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120:343–352
Matias VR, Al-Amoudi A, Dubochet J, Beveridge TJ (2003) Cryo-transmission electron microscopy of frozen-hydrated sections of Escherichia coli and Pseudomonas aeruginosa. J Bacteriol 185:6112–6118
Matricardi VR, Moretz RC, Parsons DF (1972) Electron diffraction of wet proteins: catalase. Science 177:268–270
Messaoudi C, Boudier T, Lechaire JP, Rigaud JL, Delacroix H, Gaill F, Marco S (2003) Use of cryo-negative staining in tomographic reconstruction of biological objects: application to T4 bacteriophage. Biol Cell 95:393–398
Messaoudi C, Halary S, Sorzano CO, Marco S, Boudier T (2006a) TomoJ: new tools for electron tomography. In: Proceedings of the imageJ conference, 18–19 May 2006, Luxembourg
Messaoudi C, Loubresse NGd, Boudier T, Dupuis-Williams P, Marco S (2006b) Multiple-axis tomography: applications to basal bodies from Paramecium tetraurelia. Biol Cell 98:415–425
Meyer-Ilse W, Hamamoto D, Nair A, Lelievre SA, Denbeaux G, Johnson L, Pearson AL, Yager D, Legros MA, Larabell CA (2001) High resolution protein localization using soft X-ray microscopy. J Microsc 201:395–403
Mignot J, Brugerolle G, Didier P, Bornens M (1993) Basal-body-associated macromolecules: a continuing debate. Trends Cell Biol 3:220–223
Mindell JA, Grigorieff N (2003) Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347
Mitsuoka K, Hirai T, Murata K, Miyazawa A, Kidera A, Kimura Y, Fujiyoshi Y (1999) The structure of bacteriorhodopsin at 3.0 Å resolution based on electron crystallography: implication of the charge distribution. J Mol Biol 286:861–882
Miyazawa A, Fujiyoshi Y, Unwin N (2003) Structure and gating mechanism of the acetylcholine receptor pore. Nature 423:949–955
Mobus G, Doole RC, Inkson BJ (2003) Spectroscopic electron tomography. Ultramicroscopy 96:433–451
Morgan DG, Rosier DJD (1992) Processing images of helical structures: a new twist. Ultramicroscopy 46:263–285
Moritz M, Braunfeld MB, Guenebaut V, Heuser J, Agard DA (2000) Structure of the gamma-tubulin ring complex: a template for microtubule nucleation. Nat Cell Biol 2:365–370
Navaza J (2003) On the three-dimensional reconstruction of icosahedral particles. J Struct Biol 144:13–23
Nellist PD, Chisholm MF, Dellby N, Krivanek OL, Murfitt MF, Szilagyi ZS, Lupini AR, Borisevich A Jr, Sides W, Pennycook SJ (2004) Direct sub-angstrom imaging of a crystal lattice. Science 305:1741
Nicholson WV, Glaeser RM (2001) Review: automatic particle detection in electron microscopy. J Struct Biol 133:90–101
Nickell S, Forster F, Linaroudis A, Net WD, Beck F, Hegerl R, Baumeister W, Plitzko JM (2005) TOM software toolbox: acquisition and analysis for electron tomography. J Struct Biol 149:227–234
Nickell S, Kofler C, Leis AP, Baumeister W (2006) A visual approach to proteomics. Nat Rev Mol Cell Biol 7:225–230
O’Toole ET, Giddings TH, McIntosh JR, Dutcher SK (2003) Three-dimensional organization of basal bodies from wild-type and delta-tubulin deletion strains of Chlamydomonas reinhardtii. Mol Biol Cell 14:2999–3012
Owen CH, Morgan DG, Rosier DJD (1996) Image analysis of helical objects: the Brandeis helical package. J Struct Biol 116:167–175
Penczek P, Marko M, Buttle K, Frank J (1995) Double-tilt electron tomography. Ultramicroscopy 60:393–410
Penczek P, Radermacher M, Frank J (1992) Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy 40:33–53
Penczek PA (2002) Three-dimensional spectral signal-to-noise ratio for a class of reconstruction algorithms. J Struct Biol 138:34–46
Penczek PA, Frank J, Spahn CM (2006a) A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. J Struct Biol 154:184–194
Penczek PA, Grassucci RA, Frank J (1994) The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy 53:251–270
Penczek PA, Yang C, Frank J, Spahn CM (2006b) Estimation of variance in single-particle reconstruction using the bootstrap technique. J Struct Biol 154:168–183
Penczek PA, Zhu J, Frank J (1996) A common-lines based method for determining orientations for N > 3 particle projections simultaneously. Ultramicroscopy 63:205–218
Penczek PA, Zhu J, Schröder R, Frank J (1997) Three dimensional reconstruction with contrast transfer compensation from defocus series. Scanning Microsc 11:147–154
Philippsen A, Engel HA, Engel A (2007a) The contrast-imaging function for tilted specimens. Ultramicroscopy 107:202–212
Philippsen A, Schenk AD, Signorell GA, Mariani V, Berneche S, Engel A (2007b) Collaborative EM image processing with the IPLT image processing library and toolbox. J Struct Biol 157:28–37
Potter CS, Pulokas J, Smith P, Suloway C, Carragher B (2004) Robotic grid loading system for a transmission electron microscope. J Struct Biol 146:431–440
Provencher SW, Vogel RH (1988) Three-dimensional reconstruction from electron micrographs of disordered specimens. I. Method. Ultramicroscopy 25:209–221
Radermacher M (1988) Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J Electron Microsc Tech 9:359–394
Radermacher M (1992) Weighted back-projection methods. In: Frank J (ed) Electron tomography: three-dimensional imaging with transmission electron microscope. Plenum Press, New York, pp 91–115
Radermacher M (1994) Three-dimensional reconstruction from random projections: orientational alignment via Radon transforms. Ultramicroscopy 53:121–136
Radermacher M, Rao V, Grassucci R, Frank J, Timerman AP, Fleischer S, Wagenknecht TA (1994) Cryoelectron microscopy and 3-dimensional reconstruction of the calcium release channel ryanodine receptor from skeletal-muscle. J Comput Biol 127:411–423
Radermacher M, Wagenknecht T, Verschoor A, Frank J (1987) Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc 146(Pt 2):113–136
Roseman AM (2004) FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol 145:91–99
Rosenthal PB, Henderson R (2003) Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745
Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GFX (2004) Electron crystallography reveals the structure of metarhodopsin I. EMBO J 23:3609–3620
Saad A, Ludtke SJ, Jakana J, Rixon FJ, Tsuruta H, Chiu W (2001) Fourier amplitude decay of electron cryomicroscopic images of single particles and effects on structure determination. J Struct Biol 133:32–42
Saban SD, Silvestry M, Nemerow GR, Stewart PL (2006) Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J Virol 80:12049–12059
Sali A, Glaeser R, Earnest T, Baumeister W (2003) From words to literature in structural proteomics. Nature 422:216–225
Sander B, Golas MM, Stark H (2003) Automatic CTF correction for single particles based upon multivariate statistical analysis of individual power spectra. J Struct Biol 142:392–401
Saxton WO, Baumeister W (1982) The correlation averaging of a regularly arranged bacterial envelope protein. J Microsc 127:127–138
Scheres SH, Gao H, Valle M, Herman GT, Eggermont PP, Frank J, Carazo JM (2007) Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat Methods 4:27–29
Scheres SH, Marabini R, Lanzavecchia S, Cantele F, Rutten T, Fuller SD, Carazo JM, Burnett RM, San Martin C (2005a) Classification of single-projection reconstructions for cryo-electron microscopy data of icosahedral viruses. J Struct Biol 151:79–91
Scheres SH, Valle M, Nunez R, Sorzano CO, Marabini R, Herman GT, Carazo JM (2005b) Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol 348:139–149
Schmid MB (2001) Structural proteomics: the potential of high-throughput structure determination. Trends Microbiol 10:S27–S31
Sigworth FJ (1998) A maximum-likelihood approach to single-particle image refinement. J Struct Biol 122:328–339
Skoglund U, Ofverstedt LG, Burnett R, Bricogne G (1996) Maximum-entropy three-dimensional reconstruction with deconvolution of the contrast transfer function: a test application with adenovirus. J Struct Biol 117:173–188
Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, Carazo JM, Pascual-Montano A (2004a) XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol 148:194–204
Sorzano COS, Fraga LGdl, Clackdoyle R, Carazo JM (2004b) Normalizing projection images: a study of image normalizing procedures for single particle three-dimensional electron microscopy. Ultramicroscopy 101:129–138
Sorzano COS, Jonic S, El-Bez C, Carazo JM, De Carlo S, Thevenaz P, Unser M (2004c) A multiresolution approach to orientation assignment in 3D electron microscopy of single particles. J Struct Biol 146:381–392
Sorzano COS, Marabini R, Boisset N, Rietzel E, Schroder R, Herman GT, Carazo JM (2001) The effect of overabundant projection directions on 3D reconstruction algorithms. J Struct Biol 133:108–118
Sorzano COS, Marabini R, Herman GT, Censor Y, Carazo JM (2004d) Transfer function restoration in 3D electron microscopy via iterative data refinement. Phys Med Biol 49:509–522
Sousa D, Grigorieff N (2007) Ab initio resolution measurement for single particle structures. J Struct Biol 157:201–210
Stark H, Orlova EV, Rinke-Appel J, Junke N, Muller F, Rodnina M, Wintermeyer W, Brimacombe R, van Heel M (1997) Arrangement of tRNAs in pre- and posttranslocational ribosomes revealed by electron cryomicroscopy. Cell 88:19–29
Subramaniam S, Milne JLS (2004) Three-dimensional electron microscopy at molecular resolution. Ann Rev Biophys Biomol Struct 33:141–155
Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B (2005) Automated molecular microscopy: the new Leginon system. J Struct Biol 151:41–60
Svergun DI, Koch MH (2002) Advances in structure analysis using small-angle scattering in solution. Curr Opin Struct Biol 12:654–660
Tama F, Miyashita O, Brooks CL 3rd (2004a) Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J Mol Biol 337:985–999
Tama F, Miyashita O, Brooks CL 3rd (2004b) Normal mode based flexible fitting of high-resolution structure into low-resolution experimental data from cryo-EM. J Struct Biol 147:315–326
Tao Y, Zhang W (2000) Recent developments in cryo-electron microscopy reconstruction of single particles. Curr Opin Struct Biol 10:616–622
Taylor KA, Glaeser RM (1974) Electron diffraction of frozen, hydrated protein crystals. Science 186:1036–1037
Thon F (1971) Phase contrast electron microscopy. In: Valdrè U (ed) Electron microscopy in material sciences. Academic, New York, pp 572–625
Thuman-Commike PA, Chiu W (1997) Improved common line-based icosahedral particle image orientation estimation algorithms. Ultramicroscopy 68:231–255
Thuman-Commike PA, Chiu W (2000) Reconstruction principles of icosahedral virus structure determination using electron cryomicroscopy. Micron 31:687–711
Toyoshima C, Unwin NTP (1988) Contrast transfer for frozen-hydrated specimens: determination from pairs of defocused images. Ultramicroscopy 25:279–292
Toyoshima C, Yonekura K, Sasabe H (1993) Contrast transfer for frozen-hydrated specimens II: amplitude contrast at very low frequencies. Ultramicroscopy 48:165–176
Unser M, Sorzano CO, Thevenaz P, Jonic S, El-Bez C, De Carlo S, Conway JF, Trus BL (2005) Spectral signal-to-noise ratio and resolution assessment of 3D reconstructions. J Struct Biol 149:243–255
Unser M, Trus BL, Steven AC (1987) A new resolution criterion based on spectral signal-to-noise ratios. Ultramicroscopy 23:39–51
Unwin P, Henderson R (1975) Molecular structure determination by electron microscopy of unstained crystalline specimens. J Mol Biol 94:425–440
van Heel M (1982) Detection of objects in quantum-noise-limited images. Ultramicroscopy 7:331–341
van Heel M (1984) Multivariate statistical classification of noisy images (randomly oriented biological macromolecules). Ultramicroscopy 13:165–183
van Heel M (1987) Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy 21:111–123
van Heel M, Frank J (1981) Use of multivariate statistics in analysing the images of biological macromolecules. Ultramicroscopy 6:187–194
van Heel M, Gowen B, Matadeen R, Orlova EV, Finn R, Pape T, Cohen D, Stark H, Schmidt R, Schatz M, Patwardhan A (2000) Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys 33:307–369
van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M (1996) A new generation of the IMAGIC image processing system. J Struct Biol 116:17–24
van Heel M, Schatz M (2005) Fourier shell correlation threshold criteria. J Struct Biol 151:250–262
van Marle J, Dietrich A, Jonges K, Jonges R, de Moor E, Vink A, Boon P, van Veen H (1995) EM-tomography of section collapse, a non-linear phenomenon. Microsc Res Tech 31:311–316
Velazquez-Muriel JA, Carazo JM (2007) Flexible fitting in 3D-EM with incomplete data on superfamily variability. J Struct Biol 158:165–181
Velazquez-Muriel JA, Sorzano CO, Fernandez JJ, Carazo JM (2003) A method for estimating the CTF in electron microscopy based on ARMA models and parameter adjustment. Ultramicroscopy 96:17–35
Velazquez-Muriel JA, Sorzano CO, Scheres SH, Carazo JM (2005) SPI-EM: towards a tool for predicting CATH superfamilies in 3D-EM maps. J Mol Biol 345:759–771
Vestergaard B, Sanyal S, Roessle M, Mora L, Buckingham RH, Kastrup JS, Gajhede M, Svergun DI, Ehrenberg M (2005) The SAXS solution structure of RF1 differs from its crystal structure and is similar to its ribosome bound cryo-EM structure. Mol Cell 20:929–938
Vogel RH, Provencher SW (1988) Three-dimensional reconstruction from electron micrographs of disordered specimens. II. Implementation and results. Ultramicroscopy 25:223–239
Volkmann N, Hanein D (1999) Quantitative fitting of atomic models into observed densities derived by electron microscopy. J Struct Biol 125:176–184
Walz T, Grigorieff N (1998) Electron crystallography of two-dimensional crystals of membrane proteins. J Struct Biol 121:142–161
Wang BC (1985) Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol 115:90–112
Wang HW, Nogales E (2005) An iterative Fourier-Bessel algorithm for reconstruction of helical structures with severe Bessel overlap. J Struct Biol 149:65–78
Wang L, Zhang Y, Feng J (2005) On the Euclidean distance of images. IEEE Trans Pattern Anal Mach Intell 27:1334–1339
Wang YA, Yu X, Yip C, Strynadka NC, Egelman EH (2006) Structural polymorphism in bacterial EspA filaments revealed by cryo-EM and an improved approach to helical reconstruction. Structure 14:1189–1196
White HE, Saibil HR, Ignatiou A, Orlova EV (2004) Recognition and separation of single particles with size variation by statistical analysis of their images. J Mol Biol 336:453–460
Yang C, Ng EG, Penczek PA (2005) Unified 3-D structure and projection orientation refinement using quasi-Newton algorithm. J Struct Biol 149:53–64
Zampighi GA, Zampighi L, Fain N, Wright E, Cantele F, Lanzavecchia S (2005) Conical tomography II: a method for the study of cellular organelles in thin sections. J Struct Biol 151:263–274
Zhang P, Beatty A, Milne JL, Subramaniam S (2001) Automated data collection with a Tecnai 12 electron microscope: applications for molecular imaging by cryomicroscopy. J Struct Biol 135:251–261
Zheng QS, Braunfeld MB, Sedat JW, Agard DA (2004) An improved strategy for automated electron microscopic tomography. J Struct Biol 147:91–101
Zhou ZH, Baker ML, Jiang W, Dougherty M, Jakana J, Dong G, Lu G, Chiu W (2001) Electron cryomicroscopy and bioinformatics suggest protein fold models for rice dwarf virus. Nat Struct Biol 8:868–873
Zhou ZH, Hardt S, Wang B, Sherman MB, Jakana J, Chiu W (1996) CTF determination of images of ice-embedded single particles using a graphics interface. J Struct Biol 116:216–222
Zhu J, Penczek PA, Schroder R, Frank J (1997) Three-dimensional reconstruction with contrast transfer function correction from energy-filtered cryoelectron micrographs: procedure and application to the 70S Escherichia coli ribosome. J Struct Biol 118:197–219
Zhu Y, Carragher B, Glaeser RM, Fellmann D, Bajaj C, Bern M, Mouche F, de Haas F, Hall RJ, Kriegman DJ, Ludtke SJ, Mallick SP, Penczek PA, Roseman AM, Sigworth FJ, Volkmann N, Potter CS (2004) Automatic particle selection: results of a comparative study. J Struct Biol 145:3–14
Zhu Y, Carragher B, Kriegman DJ, Milligan RA, Potter CS (2001) Automated identification of filaments in cryoelectron microscopy images. J Struct Biol 135:302–312
Ziese U, Geerts WJ, Krift TPVD, Verkleij AJ, Koster AJ (2003) Correction of autofocusing errors due to specimen tilt for automated electron tomography. J Microsc 211:179–185
Zubelli JP, Marabini R, Sorzano COS, Herman GT (2003) Three-dimensional reconstruction by Chahine’s method from electron microscopic projections corrupted by instrumental aberrations. Inverse Probl 19:933–949
Acknowledgments
We are grateful to European Commission for NoE “3D-EM” contract No. LSHG-CT-2004-502828, Region Ile-de-France for convention SESAME 2000 E 1435, program C’Nano Ile-de-France for supporting 3D cryo-electron microscopy at IMPMC, and to Institut Curie (PIC Physique du Vivant).
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented at the joint biannual meeting of the SFB-GEIMM-GRIP, Anglet France, 14–19 October, 2006.
Rights and permissions
About this article
Cite this article
Sorzano, C.O.S., Jonic, S., Cottevieille, M. et al. 3D electron microscopy of biological nanomachines: principles and applications. Eur Biophys J 36, 995–1013 (2007). https://doi.org/10.1007/s00249-007-0203-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-007-0203-x