
Abstract
Wild-type human cystatin C is directly involved in pathological fibrils formation, leading to hemorrhage, dementia and eventually death of people suffering from cerebral amyloid angiopathy. Some studies on cystatin C oligomerization have been already done but some points are still unclear. In order to learn more about this important process, we have investigated thermal and chemical (guanidine hydrochloride-induced) denaturation of human cystatin C. Studies performed using tryptophan fluorescence, calorimetry, circular dichroism and Fourier transform infrared spectroscopy demonstrate that neither chemical nor thermal denaturation of hCC are simple two-state events. One recognized intermediate form was dimeric cystatin C, whose appearance was preceded mainly by changes in the L2 binding loop. The other form occurred only in the chemical denaturation process and was characterized by partially recovered interactions maintaining the protein tertiary structure. Our studies also strongly indicate that the β-structural motif of cystatin C is directly implicated in formation of temperature-induced aggregates.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cystatins are natural protein inhibitors of cysteine proteases: proteolytic enzymes widely distributed in animals, plants and microorganisms. They are tight and reversible-binding inhibitors structurally constituting a single superfamily of evolutionarily related proteins (Rawlings and Barrett 1990). The known human members of this superfamily have been divided into three distinct families. Family 1 cystatins (cystatins A and B) are intracellular proteins containing about 100 amino acid residues (M r≈11–12 kDa) and lack disulfide bridges. Family 2 cystatins (cystatin C, D, E, F, S, SA and SN) are mainly extracellular and/or transcellular secreted proteins (M r≈13–14 kDa), possessing two intrachain disulfide bonds. The most complex Family 3 cystatins, represented by H- and L-kininogen, contain three Family 2 cystatin-like domains and are mainly intravascular proteins (Barrett et al. 1986; Grubb 2000).
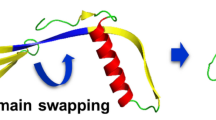
Human cystatin C (hCC) has been recognized as the most physiologically important inhibitor of cysteine proteases belonging to the papain (C1) and legumain (C13) families (Turk and Bode 1991; Grubb 2000). It is a widely distributed basic protein (pI=9.3) composed of 120 amino acids (M r=13.4 kDa) (Grubb and Lofberg 1982; Grubb 2000). The polypeptide chain of cystatin C contains three segments directly implicated in interactions with an enzyme (Bode et al. 1988). These regions include the N-terminal segment and two conserved loop-forming segments, L1 (55–59, QIVAG) and L2 (105–106, PW), which are aligned in a wedge-like fashion. The overall 3D structure of monomeric hCC has not yet been determined but a crystal structure of the dimer has been recently revealed (Janowski et al. 2001). Formed in crystallization conditions (pH 4.8), dimeric cystatin C is created through the exchange of “subdomains” (domain swapping) composed of three β-strands, β3, β4 and β5, between two protein molecules (Fig. 1). The resulting dimer consists of two identical subunits, each with a large five-stranded antiparallel β-sheet wrapped around a long α-helix, which resembles a well-known general fold of chicken cystatin C (Bode et al. 1988).

Structure of the dimeric form of human cystatin C
Studies of hCC self-association and aggregation processes have been performed because this protein is directly involved in the formation of pathological fibrillar aggregates which deposit in the brain arteries of elderly persons, causing cerebral amyloid angiopathy. Some studies of the cystatin C aggregation process have been already undertaken (Ekiel and Abrahamson 1996, Ekiel et al. 1997), showing that in some conditions (low pH, high temperature or the presence of a denaturing agent) the protein exists in a dimeric form. However, it is still not clear what kind of changes precede the dimer formation or what happens to the protein structure after the dimer disruption. To address these questions, we followed changes induced in the protein structure by temperature or a denaturing agent (guanidine hydrochloride, Gdn.HCl) by means of differential scanning calorimetry (DSC) and a number of spectroscopic methods, including circular dichroism (CD), fluorescence and Fourier transform infrared (FT-IR) spectroscopy.
Materials and methods
Cystatin C was kindly provided by Professor Anders Grubb from the University Hospital of Lund, Sweden. The native protein was the monomer, which was checked by agarose gel electrophoresis (protein concentration in the sample was 75 μM).
The protein solutions for all experiments apart from FT-IR were prepared by dilution of cystatin C lyophilized powder in 50 mM phosphate buffer (pH 6.6) containing 0.1 M NaCl. The protein concentration in all samples was determined on the basis of the absorbance at 280 nm (ε280=14,800 M−1 cm−1).
Circular dichroism
All CD experiments were carried out on an Aviv 62 spectropolarimeter.
Chemical denaturation
Spectra were recorded at room temperature (22 °C) for two spectral ranges: 190–260 nm (far-UV) and 240–330 nm (near-UV). The aromatic spectral range for solutions with a Gdn.HCl concentration higher than 2.25 M was narrowed to 250–310 nm. Cuvettes of 1 mm and 5 mm path length were used for the far- and near-UV CD measurements, respectively. All spectra were recorded with 0.2 nm resolution. The protein concentration was 37.6 μM.
Thermal denaturation
Spectra were recorded at 222 nm in a cuvette of 0.5 cm path length. Data were collected in the range from 1 to 99 °C with intervals of 2 °C and 5 °C in a heating and cooling cycle, respectively. The sample was equilibrated at each temperature for 1 min. The protein concentration was 42.9 μM.
Fluorescence spectroscopy
Fluorescence measurements were performed using a Perkin-Elmer LS-50B spectrofluorometer. The protein concentration was 9.4 μM in both chemical and thermal denaturation experiments. The excitation wavelength was set at 295 nm. The emission spectrum of tryptophan was recorded in the range from 310 to 500 nm.
Chemical denaturation
Cystatin C solutions with different amounts of Gdn.HCl were incubated at room temperature for 18–24 h. The exact concentration of the denaturing agent in the sample was determined refractometrically. Fluorescence spectra were recorded at 22 °C. The spectral slit width was 6.0 nm for both excitation and emission wavelengths.
Thermal denaturation
The spectra were recorded in a heating (from 5 to 80 °C) and then in a cooling (from 80 to 2 °C) cycle. The equilibrium time for each temperature was 15 min. N-Acetyltryptophanamide was used as a reference and studied in the same buffer and temperature conditions as the protein. The excitation wavelength for N-acetyltryptophanamide was 275 nm. The slit width was set at 6.0 nm (for cystatin C) or 3.5 nm (for N-acetyltryptophanamide).
FT-IR spectroscopy
Infrared spectra were recorded for cystatin C solution in heavy water and for solid cystatin C using a Bruker IFS-66 spectrometer equipped with a deuterated triglycine sulfate detector. In the heating cycle, data were collected from 35 to 90 °C with intervals of 15 °C. Similar intervals were applied in the cooling cycle for solid cystatin C, while for the protein solution only a spectrum at 44 °C was recorded. At each temperature the samples were equilibrated for 15 min. From 500 to 1024 scans at a resolution of 2 cm−1 were averaged in each measurement.
A sample for measurements in the solid state was prepared by dissolving cystatin C in water (2% w/v), placing 10 μL of the solution on the CaF2 plate and evaporating the solvent. Deuterated cystatin C was prepared by incubation of the protein in heavy water and then lyophilization. Completeness of the proton–deuterium exchange was judged by disappearance of the amide II band corresponding to the N–H bending vibrational mode. The protein concentration was 2% (w/v) and the path length 25 μm. Spectra of the solvent were acquired under the same scanning and temperature conditions.
Data processing was done using GRAMS/32 version 5.22 software (Galactic Enterprises). Spectral contributions from residual water vapor were eliminated using a set of water vapor spectra. The tyrosine absorption band at 1516 cm−1 was used as a reference point for baseline correction (Mohney et al. 2000). Correct subtraction of the solvent spectra was judged to yield an approximately flat baseline in the region where heavy water has strong absorption (near 1220 cm−1). A nine-point Savitzky–Golay smoothing function was applied to calculate the second derivative. Deconvolution of the spectra was done in the spectral range 1500–1800 cm−1, using γ-factor 5.5. The deconvoluted spectra were fitted with gaussian band profiles. The positions and number of the components, which were used as an input file for the curve-fitting function, were obtained from both the second derivative and the deconvoluted spectra. The quality of the fitting was estimated by standard deviation.
Differential scanning calorimetry
DSC experiments were carried out on a Microcal MC-2 scanning microcalorimeter. Samples were dialyzed against phosphate buffer and then centrifuged. Aliquots of the final dialysis buffer were used for reference. The protein concentration was 0.207 mM. The sample and reference solutions were kept during experiments under a N2 atmosphere (2 atm) to avoid solvent evaporation and bubble formation at higher temperatures. The heating rate was 1 °C per minute. The obtained data were analyzed using Microcal Origin software package. Cubic splines interpolation was used for baseline correction. The calorimetric enthalpy (ΔH cal) was estimated from the area under the thermometric curve. The van’t Hoff enthalpy (ΔH vH) was calculated on the basis of the equation: ΔH vH=4RT 2 c p,max/ΔH cal, where c p,max is the maximal heat capacity, R is the gas constant and T is the temperature.
Results and discussion
Chemical denaturation
Changes caused by Gdn.HCl in the environment of the cystatin C tryptophan residue (Trp106 from the L2 binding loop) were monitored by means of fluorescence spectroscopy. The spectrum of the native protein excited at 295 nm has an emission maximum at 352 nm. Increasing amounts of Gdn.HCl caused a gradual red shift of this maximum, indicating transition of the tryptophan residue to a more exposed position (Irace et al. 1981; Freskgård et al. 1998) (Fig. 2A). As expected, the change was relatively small (about 2 nm) because Trp106 is substantially exposed to the solvent already in the native protein molecule (Grzonka et al. 1992).

Guanidine hydrochloride denaturation of wild-type cystatin C [50 mM phosphate buffer containing 0.1 M NaCl, pH 6.6, protein concentration 9.4 μM (A, B) or 37.6 μM (C), room temperature] followed by: A the tryptophan (Trp106) emission maximum as a function of Gdn.HCl concentration; B the tryptophan fluorescence intensity at λ=356 nm (F 356=fluorescence intensity at different Gdn.HCl concentrations; F 0=fluorescence intensity at 0 M Gdn.HCl); λexc=295 nm for both A and B. C Changes in the vicinity of aromatic residues followed by changes in the CD signal recorded at 275 nm
Owing to the quenching capability of the solvent, the tryptophan exposition should be accompanied by a decrease in its fluorescence intensity (Goto and Fink 1989; Lakowicz 1999). Figure 2B shows, however, that up to 2.0 M Gdn.HCl the Trp106 fluorescence intensity gradually increases. This suggests that quenching of the tryptophan fluorescence by the solvent is somehow compensated, probably by a growing distance between Trp106 and those parts of the molecule which contain intrinsic energy quenchers: the side chain of lysine, histidine or methionine residues, or a disulfide bridge (Havel et al. 1988).
Gradual separation of Trp106 from the moieties capable of quenching its fluorescence was stopped twice during the denaturation process. One break appeared between Gdn.HCl concentrations of 0.5 and 1.0 M (Fig. 2B) and reflected the existence of a first intermediate on the cystatin C denaturation path. Most probably this intermediate was the dimeric form of cystatin C, since such a form was recognized at the same Gdn.HCl concentration in studies done previously by Ekiel and Abrahamson (1996).
Existence of the intermediate in the range 0.5–1.0 M Gdn.HCl was corroborated also by our near-UV CD experiments (Fig. 2C), as well as far-UV CD spectra recorded at 222 nm (Fig. 3). The curves depicted in Fig. 3 show how Gdn.HCl influences cystatin C secondary structure elements: the α-helix (222 nm) and β-sheet (217 nm). The curves clearly demonstrate that the denaturation process did not proceed in exactly the same way in the α-helical and β-structural regions of the cystatin C molecule. Structural transformations of the β-sheet motif are monotonous with only one transition point at about 1.5 M Gdn.HCl, whereas on the curve recorded at 222 nm there is also an inflexion indicating existence of the intermediate.

Gdn.HCl-induced changes in the CD signal, reflecting transformations in the α-helical (222 nm) and β-structural regions (217 nm) of wild-type cystatin C (same buffer conditions as in Fig. 2; protein concentration 37.6 μM)
It seems quite significant that existence of the intermediate form was clearly visible in every structural motif studied (Fig. 2B, Fig. 2C, Fig. 3 at 222 nm) except the β-sheet. The undisturbed continuity of the conformational transformations in the β-region during the whole denaturation process may indicate that the β-structure is in some way responsible for both the intermediate (the dimer) formation and disruption. An initial decrease in interactions maintaining the native fold in the β-region might cause a gradual drop in the monomer stability, forcing the cystatin C molecule to adopt a more stable dimeric form. Subsequent changes within the β-structure might decrease also the dimer stability and eventually lead to its disruption.
This supposition as to the role of the β-sheet structural motif in the cystatin C dimerization process has been confirmed by recently conducted crystallographic studies of hCC (Janowski et al. 2001). The protein, monomeric before crystallization, was obtained in the crystal as the dimer, formed through exchange of the β-structure domains between two molecules. The mechanism of the process comprised disruption of the native interactions between the β2 and β3 strands of each molecule and creation from them of the unusually long intermolecular β-sheet structure (Fig. 1).
It is difficult to say at this stage whether the same mechanism was engaged in the formation of dimers during our chemical denaturation process. It is, however, possible that initially observed changes in the Trp106 position (Fig. 2B) were a preparation for the domain swapping event, since the L2 loop is adjacent to the L1 binding loop which serves as a hinge in the swapping of the β-sheet domains. Moreover, Ekiel and co-workers’ NMR study of cystatin C dimers and monomers (Ekiel et al. 1997), which speaks in favor of the domain swapping mechanism, also pointed out the L2 binding loop as the region most (although not significantly) affected in the denaturation process.
The dimer was not the only intermediate formed on the pathway toward cystatin C total denaturation. The second intermediate form occurred between 2.5 and 4.0 M Gdn.HCl and was visible on the fluorescence intensity curve (Fig. 2B) as well as on the near-UV CD spectra (Fig. 2C).
The initial decrease and subsequent increase which took place in the tryptophan fluorescence intensity in the range 2.5–4.0 M Gdn.HCl (Fig. 2B) could be explained, as previously, as being caused by interactions of the tryptophan residue with the intrinsic quenching groups. We found, however, that it could also be the effect of alterations in the efficiency of tyrosine→tryptophan energy transfer (Havel et al. 1988). This last explanation was indicated by changes in the tyrosine emission intensities, which in that range of denaturant concentrations proceeded in a way exactly opposite to that of tryptophan (data not shown). It is then possible that the decrease in the tryptophan emission intensity reflected the gradual separation of the L2 loop from the protein segments containing tyrosine residues. There are four such segments in the cystatin C molecule: the α-helix and the β2, β3 and β4 strands. The structures of chicken cystatin C as well as the dimeric form of its human counterpart suggest that tyrosine residues from the α-helix and the β2 strand are too far away to interact with Trp106. The most probable source of the changes seems to be the growing distance between the L2 binding loop and tyrosine residues from the β3 and/or β4 strands, which evokes once again the β-structure as a motif implicated somehow in the structural transformations observed during the cystatin C denaturation process.
Alterations in the CD signal in the range 2.5–4.0 M Gdn.HCl (Fig. 2C) could be connected with tyrosine movements as well, since the near-UV region provides insight into conformational transformations in the proximity of aromatic amino acids (Carey and Surewicz 1996). In the case of the CD experiment, however, the situation is quite complicated as there are a lot of different chromophores in the cystatin C molecule which could give rise to changes in the ellipticity. However, tyrosine is an aromatic residue and although it might not be the only source of changes in the CD signal, it is possible that transfer of some tyrosine residues to a more hydrophobic inner location contributed, at least in part, to the temporary increase in the negative ellipticity and formation of the second intermediate form on the cystatin C denaturation path.
A form similar to this second intermediate was observed in almost the same denaturant concentration range (3.0–4.0 M Gdn.HCl) in the chemical denaturation process of chicken cystatin C (Björk and Pol 1992). Moreover, NMR studies of a cystatin C L68Q mutant allowed detection of a second intermediate in a pH-induced denaturation process (Gerhartz et al. 1998). These results go in concert with ours in indicating that the denaturation process of cystatin C is a quite complicated event with more than one transition point occurring.
Thermal denaturation
Alterations in the fluorescence of N-acetyltryptophanamide were studied in order to distinguish between a temperature influence on the indole ring and on the cystatin C conformation. The decreases in the fluorescence intensity of the protein and the reference compound were almost identical up to 25 °C (results not shown). At higher temperatures, divergence between them gradually grew, demonstrating the occurrence of rearrangements in the vicinity of the protein tryptophan residue (Trp106 in the L2 binding loop).
At the beginning of the denaturation process, a decrease in the Trp106 emission intensity took place simultaneously with a transfer of the residue to a more buried position (Fig. 4, inset). However, above 35 °C the emission maximum initially did not change at all, then started to shift to higher wavenumbers and both these events were accompanied by a monotonic decrease in the fluorescence intensity (Fig. 4). Independent changes of the emission maximum and intensity suggested that the quenching activity of the solvent was not the main reason for the tryptophan fluorescence decrease. One should rather assume that this decrease was caused by more tight interactions of the Trp106 residue with the intrinsic energy quenchers as a result of the greater compactness of the protein molecule.

Temperature dependence of the center of spectral mass (<ν>) of Trp106 fluorescence, where <ν>=∑(ν i F i )/∑F i (Ruan et al. 1997), ν i =wavenumber, F i =fluorescence intensity. Inset: temperature dependence of the tryptophan fluorescence maximum. Protein concentration 9.4 μM; λexc=295 nm; buffer conditions the same as in Fig. 2
Such a surprising direction of changes leading to a more compact protein structure at the beginning of the denaturation process was also observed in our CD experiments. CD spectra of hCC recorded at 222 nm enable detection of structural changes proceeding in the helical region of the cystatin C molecule. The results presented in Fig. 5 show that structural transformations of this segment initially led to increased rigidity of the α-helix. This is in contrast to the chemical denaturation process, where initial structural transformations induced loosening of the protein structure. The structural changes at the beginning of both chemical and thermal processes were relatively small; nevertheless, their opposite direction was clearly noticeable.

Temperature-induced changes in cystatin C α-helical regions monitored by CD at 222 nm (buffer conditions as in Fig. 2; protein concentration 42.9 μM)
Another difference between the thermal and chemical denaturation events was the form undergoing main transformations in the unfolding process. In the chemical denaturation process, dimer disruption preceded substantial alterations in the protein structure. In the thermal event, a quick drop in the negative ellipticity, reflecting significant lost of the protein native structure, appeared above 65 °C (Fig. 5), when, as previous investigations of Ekiel and Abrahamson (1996) have demonstrated, hCC should exist in its dimeric form. So, in the thermal process the dimeric form of cystatin C was the moiety participating in the main structural transformations.
This statement was also corroborated by the results of our calorimetric studies. The calorimetric scans of cystatin C showed one distinct peak with a maximum at about 81 °C (Fig. 6, solid line). The ratio between the heat effect estimated from the area under the peak and the van’t Hoff enthalpy calculated from the respective equation (see Materials and methods) was equal to 1.89, which distinctly points to the oligomeric (dimeric) nature of the moiety undergoing thermal unfolding. The thermogram obtained in the second experiment conducted for the same sample (Fig. 6, dashed line) revealed no changes in the heat capacity, indicating that the end point of the unfolding process had been achieved during the first heating cycle. The irreversibility of the cystatin C thermal denaturation process demonstrated by the calorimetric studies agrees with the results of our fluorescence (Fig. 4) as well as CD studies (Fig. 5), which also indicated irreversibility of the temperature-induced denaturation of hCC.

Experimental (solid line: first heating; dashed line: reheating) and theoretical (dotted line) endotherms for the thermal denaturation of cystatin C (buffer conditions as in Fig. 2; protein concentration 0.207 mM; heating rate 1 °C/min)
Important information about the very last step in the cystatin C denaturation pathway was given by FT-IR studies. Structural information in the FT-IR method is predominantly derived from analysis of the so-called amide bands, particularly the amide I band (1600–1700 cm−1). This band comprises mainly the peptide backbone C=O stretching vibration and usually has a broad contour which is composed of several overlapping bands due to various protein segments with different secondary structure (Fabian et al. 1992). Deconvolution and derivation of the amide band make it possible to distinguish between the individual components and assign each of them to a specific conformational type (Casal et al. 1988; Venyaminov and Kalnin 1990).
The native cystatin C amide I band (spectrum recorded at 35 °C) was composed of at least 12 components (results not shown). Curve-fitting results allowed estimation of the quantitative contributions of each band to the amide I band contour. Calculations based on the cystatin C spectrum obtained at 35 °C indicated that the native protein conformation consisted of 45% β-sheet, almost 15% α-helix and about 22% of turns (Table 1). The rest of the protein chain was mainly disordered. These results conformed well with the results obtained from the crystallographic studies of cystatin C: the percentage content of β-structure and α-helix in the monomeric form of cystatin C (extracted from the dimer) was estimated as about 47% and 16%, respectively (Janowski et al. 2001).
The amide I bands obtained at 50 and 75 °C did not differ much from that recorded at 35 °C (results not shown). The band obtained at 75 °C was only a little broader and less intensive than the bands recorded at lower temperatures. Distinct changes in the secondary structure of cystatin C appeared between 75 and 90 °C. The amide I band contour recorded at 90 °C was significantly different than that obtained at 75 °C. The change was even more visible in the second-derivative spectra calculated for these amide bands (Fig. 7). The second-derivative spectrum of the amide I band obtained at 90 °C revealed two striking features. The first one was a substantial decrease in the intensity of a lobe representing the normal β-sheet structural motif (~1630 cm−1); the second was the appearance of two new large lobes, at 1617 and 1684 cm−1. According to the literature, these new bands represented β-sheet aggregates (Banecki et al. 1992; Oberg and Fink 1998; Mohney et al. 2000). The percentage content of these aggregates in the cystatin C molecule reached 25% (Table 1). Taking into account that the appearance of these aggregates was accompanied by almost disappearance of the normal β-sheet structure, it seems very likely that the β-strands were directly involved in the formation of these aggregates.

The second-derivative of cystatin C FT-IR spectra recorded at 75 °C (dashed line) and 90 °C (solid line). Spectra were recorded for 2% (w/v) cystatin C in heavy water; path length 25 μm
The amide I band and its second-derivative spectrum obtained for cystatin C solution cooled to 44 °C were essentially the same as the ones obtained at 90 °C (results not shown). It confirmed once more the irreversibility of the human cystatin C thermal denaturation process.
Besides studies on cystatin C in heavy water, trials were undertaken to observe the denaturation process for solid cystatin C, since the spectrum without a solvent contribution would be much easier to interpret. It was found that evaporation of the solvent did not disturb the protein structure, as was previously reported (Oberg and Fink 1998). However, even intensive heating of solid cystatin C was not able to induce changes in its native structure (results not shown). It gave rise to the conclusion that although it is possible to determine the protein structure on the basis of FT-IR studies in the solid state, studies of proteins’ conformational transformations give reasonable results only for their solutions.
Conclusions
The obtained results demonstrate that thermal and chemical denaturation processes of wild-type hCC proceed in a different manner but neither are simple two-state events. One intermediate form – the only one in the thermal process and one of two in the chemical event – is dimeric cystatin C. The appearance of the dimer is preceded by transformations in the protein tertiary structure, while the secondary arrangement stays almost unchanged.
The β-structure seems to be directly involved in the dimer formation and disruption. This statement stands in line with the results obtained in X-ray studies of hCC (Janowski et al. 2001), which revealed that the dimers result from the three-dimensional domain swapping and that the dimer interface is created from the β2 and β3 strands contributed by two hCC monomers. Dimer disruption in the thermal denaturation is followed by aggregate formation, in which process the β-strands are also engaged unambiguously.
It is not stated if a domain swapping mechanism is responsible for the protein oligomerization and amyloid formation. Also it has not been proved yet whether our β-aggregates have the fibrous amyloidogenic structure. However, the fact that all studies unanimously point to the β-structure as being directly implicated in cystatin C oligomer formation strongly suggests that the same structural motif and the same procedures are involved in the protein pathological transformations in vivo.
Abbreviations
- Gdn.HCl:
-
guanidine hydrochloride
- hCC:
-
human cystatin C
References
Banecki B, Żylicz M, Bertoli E, Tanfani F (1992) Structural and functional relationships in DnaK and DnaK756 heat-shock proteins from Escherichia coli. J Biol Chem 267:25051–25058
Barrett AJ, Fritz H, Grubb A, Isemura S, Järvinen M, Katunuma N, Machleidt W, Müller-Esterl W, Sasaki M, Turk V (1986) Nomenclature and classification of the proteins homologous with the cysteine-proteinase inhibitor chicken cystatin. Biochem J 236:312–316
Björk I, Pol E (1992) Biphasic transition curve on denaturation of chicken cystatin by guanidinum hydrochloride. FEBS Lett 299:66–68
Bode W, Engh R, Musil D, Thiele R, Huber R, Karshnikov A, Brzin J, Kos J, Turk V (1988) The 2.0 Å X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J 7:2593–2599
Carey PR, Surewicz WK (1996) Spectroscopic and calorimetric methods for characterizing proteins and peptides. Protein Eng Des 231–263
Casal HL, Köhler U, Mantsch HH (1988) Structural and conformational changes of β-lactoglobulin B: an infrared spectroscopic study of the effect of pH and temperature. Biochim Biophys Acta 957:11–20
Ekiel I, Abrahamson M (1996) Folding-related dimerization of human cystatin C. J Biol Chem 271:1–8
Ekiel I, Abrahamson M, Fulton DB, Lindhal P, Storer AC, Levadoux W, Lafrance M, Labelle S, Pomerleau Y, Groleau D, LeSauteur L, Gehring K (1997) NMR structural studies of human cystatin C dimers and monomers. J Mol Biol 271:266–277
Fabian H, Naumann D, Misselwitz R, Ristau O, Gerlach D, Welfle H (1992) Secondary structure of streptokinase in aqueous solution: a Fourier transform infrared spectroscopic study. Biochemistry 31:6532–6538
Freskgård P-O, Petersen LC, Gabriel DA, Li X, Persson E (1998) Conformational stability of factor VIIa: biophysical studies of thermal and guanidine hydrochloride-induced denaturation. Biochemistry 37:7203–7212
Gerhartz B, Ekiel I, Abrahamson M (1998) Two stable unfolding intermediates of the disease-causing L68Q variant of human cystatin C. Biochemistry 37:17309–17317
Goto Y, Fink AL (1989) Conformational states of B-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28:945–952
Grubb A (2000) Cystatin C: properties and use as diagnostic marker. Adv Clin Chem 35:63–99
Grubb A, Lofberg H (1982) Human gamma-trace, a basic microprotein: amino acid sequence and presence in the adenohypophysis. Proc Natl Acad Sci USA 79:3024–3027
Grzonka Z, Kasprzykowski F, Ołdziej S, Wiczk W, Grubb A (1992) Recent developments in the chemistry and biology of cystatins. In: Yanaihara N (ed) Peptide chemistry 1992. Escom, Leiden, pp 243–246
Havel HA, Kauffman EW, Elzinga PA (1988) Fluorescence quenching studies of bovine growth hormone in several conformational states. Biochim Biophys Acta 955:154–163
Irace G, Parlato G, Servillo L, Colonna G (1981) Tryptophanyl fluorescence heterogeneity of apomyoglobins. Correlation with presence of two distinct structural domains. Biochemistry 20:792–799
Janowski R, Kozak M, Jankowska E, Grzonka Z, Grubb A, Abrahamson M, Jaskólski M (2001) Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat Struct Biol 8:316–320
Lakowicz JR (1999) Principles of fluorescence spectroscopy, 2nd edn. Kluwer/Plenum, New York, chap 8
Mohney BK, Petri ET, Uvarova V, Walker GC (2000) Infrared absorption and ultraviolet-circular dichroism spectral studies of thermally induced unfolding of apomyoglobin. Appl Spectrosc 54:9–14
Oberg KA, Fink AL (1998) A new attenuated total reflectance Fourier transform infrared spectroscopy method for the study of proteins in solution. Anal Biochem 256:92–106
Rawlings ND, Barrett AJ (1990) Evolution of proteins of the cystatin superfamily. J Mol Evol 30:60–71
Ruan K, Lange R, Bec N, Balny C (1997) A stable partly denatured state of trypsin induced by high hydrostatic pressure. Biochem Biophys Res Commun 239:150–154
Turk V, Bode W (1991) The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett 285:213–219
Venyaminov SY, Kalnin NN (1990) Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers 30:1243–1257
Acknowledgements
We wish to thank Bogdan Banecki for the DSC studies and are also grateful to Gabriela Nowak-Wiczk and Joanna Hebanowska for their technical assistance. This work was supported by the State Committee for Scientific Research (KBN, Poland).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jankowska, E., Wiczk, W. & Grzonka, Z. Thermal and guanidine hydrochloride-induced denaturation of human cystatin C. Eur Biophys J 33, 454–461 (2004). https://doi.org/10.1007/s00249-003-0384-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-003-0384-x