
Abstract
Bats are important zoonotic reservoirs for many pathogens worldwide. Although their highly specialized ectoparasites, bat flies (Diptera: Hippoboscoidea), can transmit Bartonella bacteria including human pathogens, their eco-epidemiology is unexplored. Here, we analyzed the prevalence and diversity of Bartonella strains sampled from 10 bat fly species from 14 European bat species. We found high prevalence of Bartonella spp. in most bat fly species with wide geographical distribution. Bat species explained most of the variance in Bartonella distribution with the highest prevalence of infected flies recorded in species living in dense groups exclusively in caves. Bat gender but not bat fly gender was also an important factor with the more mobile male bats giving more opportunity for the ectoparasites to access several host individuals. We detected high diversity of Bartonella strains (18 sequences, 7 genotypes, in 9 bat fly species) comparable with tropical assemblages of bat-bat fly association. Most genotypes are novel (15 out of 18 recorded strains have a similarity of 92–99%, with three sequences having 100% similarity to Bartonella spp. sequences deposited in GenBank) with currently unknown pathogenicity; however, 4 of these sequences are similar (up to 92% sequence similarity) to Bartonella spp. with known zoonotic potential. The high prevalence and diversity of Bartonella spp. suggests a long shared evolution of these bacteria with bat flies and bats providing excellent study targets for the eco-epidemiology of host-vector-pathogen cycles.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Bats (Chiroptera) are the second largest order of mammals, with the number of extant species over 1250 [1]. The order Chiroptera contains two suborders Yinpterochiroptera and Yangochiroptera; these include biologically and ecologically diverse species [2]. Both suborders have wide distribution, with a primarily tropical and temperate (Yinpterochiroptera), but even arctic, representatives (Yangochiroptera, [2]). Bats host a number of arthropod ectoparasites, like mites (Acari), ticks (Ixodida), fleas (Siphonaptera), and flies (Diptera). One of their specialized ectoparasites are the hippoboscoid flies (Hippoboscoidea: Nycteribiidae and Streblidae [3]), mostly flightless flies occurring only on bats [3, 4]. Hippoboscoid flies are semi-permanent parasites, spending their entire life on the host’s body, exclusively females leaving the host for a short time when depositing the third stage larvae ready to pupate in the environment. These flies are considered to act as vectors of pathogens [5]. Bats themselves are important zoonotic reservoirs for a number of pathogens. Among these, viruses are especially important and well documented [6], while our knowledge on bacteria [7] and piroplasms [8,9,10] are limited. This is even more pronounced for arthropod-borne bacteria, for which there are only a handful of studies, reporting primarily pathogens from Rickettsia [11], Borrelia [12], and Bartonella genera [13, 14]. While the incidence of Bartonella in bat species has been known from some time [15], it has only recently attracted research interest. As such, recent surveys linked the presence of pathogenic Bartonella spp. in bats to molecular detection of this pathogen in ectoparasitic Nycteribiidae flies [16, 17], suggesting the importance of these hippoboscoid flies as vectors of Bartonella spp. [5].
Bartonella spp. are facultative intracellular parasites, which are developing in erythrocytes and endothelial cells of a number of mammalian species. These bacteria may cause chronic intra-erythrocyte infections, with a complex of humoral, neurologic, and ocular manifestations in humans and several domestic mammals. Infection in reservoir hosts is usually without clinical signs [18]. Recent studies suggest that Bartonella evolved in mammals in such a way that they became highly adapted to their host species or group, currently most known Bartonella spp. being host restricted [14, 19]. With the rapid emergence of newly described Bartonella infections worldwide [20], knowledge on the reservoirs, vectors, host-ranges, and transmission dynamics is needed for adequate surveillance of possible zoonotic Bartonella strains. Bats and their associated Nycteribiidae flies are suggested to be important reservoirs for diverse Bartonella spp. in Africa [21,22,23], Asia [24, 25], and the New World [26, 27]. Bartonella strains were already indicated in bat ectoparasites from the region [28], but taking into account the overall scarcity of studies in Europe [16, 29], as well the continuous increase of cave-inhabiting bat populations all over Europe [30], this study intends to widen our knowledge on Bartonella spp. related to insectivorous bats in Central and Eastern Europe.
The aim of the present work was to assess, by PCR and sequencing, the prevalence and diversity of Bartonella strains in nycteribiid flies collected from bats occurring naturally in Hungary and Romania, and to study the diversity of Bartonella spp., while using characteristics of host and vector ecology to explain this diversity among hosts and habitats. In order to reach this, we molecularly identified Bartonella sequences from parasitic flies and compared them to sequences deposited in GenBank and evaluated the importance of vertebrate host and insect vector ecology for the presence and prevalence of these bacteria.
Methods
Sampling Sites
Samples analyzed in this study were collected in Central and Eastern Europe, in Hungary and Romania. Hippoboscoid flies (Hippoboscoidea: Nycteribiidae) were collected from bats at 38 different bat capture sites distributed in the Carpathians and the Dobrogean Plateau (Romania) and in various, mainly mountainous parts of Hungary (Fig. 1). Study areas included roosting sites localized in buildings, caves and mine galleries, drinking and foraging areas, as well sites used for mating (swarming sites). Dates for bat captures ranged from 2007 to 2015.

Geographical distribution of sampling locations for Nycteribiidae flies used in this study
Collection of Bat Flies
Bat flies were collected from individual bats, using forceps or with the help of a Fair Isle Apparatus [3]. Bats were identified to species based on morphological keys, with sex and age identified (based on tooth-wear and metacarpal joint ossification) for all specimens [31]. All ectoparasites from each individual bat were collected to allow prevalence data to be calculated. Preservation and long-term storage of bat flies was in 70 or 87% ethanol in separate vials (one vial per bat host). Identification of bat flies was based on morphological characteristics [32, 33].
To assess the importance of vector ecology, we assigned each bat fly species to one group (either mono-, oligo-, or polyxenous), based on the host specificity of the particular Nycteribiidae species [3]. Bat species were also grouped according to their affinity to a particular roost type in the non-hibernating period, thus creating three groups: (1) cave (including mines), (2) building, or (3) tree specialists [31].
DNA Extraction, PCR, and Sequencing
DNA from bat flies was extracted with ammonium hydroxide as described previously [34]. Bat flies were tested individually for presence of Bartonella spp. using a conventional PCR assay, targeting the citrate synthase gene gltA using primer sequences BhCS.781p (5’-GGGGACCAGCTCAtGGTGG) and BhCS.1137n (5’-AATGCAAAAAGAACAGTAAACA), yielding amplification products of approximately 380 base pairs [35]. For amplification, an initial denaturation step at 95 °C for 20 s (3 cycles) was followed by 3 cycles of annealing at 55 °C for 20 s, 3 cycles at 53 °C for 20 s, and 35 cycles of 51 °C for 20 s. The final extension was performed at 72 °C for 1 min. PCR products were electrophoresed and visualized in a 1.5% agarose gel. Both strands of PCR products were sequenced with the Sanger method (BaseClear, Leiden, the Netherlands), using the same forward and reverse primers as in the conventional PCR. Trimming, manual editing, aligning, and cluster analyses of Bartonella sequences were performed in Bionumerics 7.1. (Applied Math, Belgium) together with Bartonella reference sequences available in GenBank.
Phylogenetic Analyses and the Visualization of the Host-Parasite-Pathogen Network
DNA sequences from this study and from the GenBank were aligned and clustered using pairwise and multiple alignments applying Neighbor-joining method in Bionumerics 7.1. The Jukes and Cantor model was used for the rate of nucleotide substitution. Bootstrap values were calculated by the analysis of 1000 replicates. We delineated seven Bartonella clusters by visually inspecting a phylogenetic tree (supplementary Fig. S1. For presentation of the parasite network, we used the “bipartite” package in R, function “plotweb” [36].
Statistical Analyses
For assessing microparasite (Bartonella spp.) species richness, we calculated the Shannon index (H) for each bat and bat fly species, as well as for the ecologic group of bat flies (e.g., groups made by mono-, oligo-, or polyxenous species) or bats (cave-, tree-, or building-dwellers).
We fitted GLMM’s in the statistical computing environment R version 3. 3. 2 [37]. We used binomial generalized linear mixed models (GLMM) with the package “lme4” [38], using the function “glmer.” Output variable was Bartonella spp. presence/absence. Random variables were bat specimens, since some of them provided multiple samples. In the model with bat species as input variable, we used roosting places, bat id’s, and bat fly species as nested random variables. In the model with bat fly species as input variable, we used the following nested random variables: roosting places, bat species, and individual bat id’s. In the model with bat- and bat fly gender and roosting places as input variables, we used bat species, bat id’s, and bat fly species as nested random variables.
We assume that one Bartonella genotype might be widespread among samples from a particular condition, e.g., a specific combination of a bat species and one ectoparasite species. We tested this possibility using a multinomial model in which the fractions of the seven genotypes are proportional to the total sample sizes per genotype. We then calculated the probability: observing a specific genotype as frequently as or more frequently than actually observed in the sample. The probability (i.e., p values) less than 0.05 were considered significant support for selective distribution. In addition, rarity of a genotype is evaluated by calculating the probability: observing a specific genotype as frequently as or less frequently than actually observed.
Next, likely explanations for the excess or deficit of a genotype involve the bat family, bat species, ectoparasite, and the country. We evaluated these possibilities, by fitting a multinomial model to the dataset in which the fractions of the seven genotypes are specific to the bat family, bat species, ectoparasite, or country. Fit was performed by maximizing the log-likelihood, and the model for which the Akaike Information Criterion is the lowest is designated to be the best-fit model. All computation was performed using the software Mathematica version 11.0.1 (Wolfram Research Inc., Champaign, IL).
Availability of Data and Material
All the data from this study is freely available (upon registration) on the www.geo-parasite.orgwebpage.
Results
Bats, Bat Flies, and Bartonella Prevalences
Altogether, 544 individual bat flies belonging to 10 species were analyzed. These were collected from 305 individuals of 14 bat species (Hungary, 197 individuals of 10 bat fly species; Romania, 346 individuals of 7 bat fly species, Table 1). A number of 158 bat flies were positive for Bartonella spp. DNA (29.1%; CI 25.3–33.1), from which 148 samples were successfully characterized representing 18 unique Bartonella sequences. Bartonella spp. was detected and sequenced in 9 out of 10 Nycteribiidae fly species, collected from 11 out of the 14 bat species studied (Supplementary Table S1).
Association of Bartonella Strains with Bat Flies
Bartonella spp. sequences were obtained from nine nycteribiid species: Basilia nana, B. nattereri, Nycteribia kolenatii, N. pedicularia, N. schmidlii, N. vexata, Penicillidia conspicua, Pe. dufouri, and Phthiridium biarticulatum, while we were unable to detect any in N. latreillii. The distribution of Bartonella spp. in individual bat fly species was not linear, with certain nycteribiids hosting higher prevalence or diversity of individual Bartonella sequences. Polyxenous bat flies had the lowest prevalence (18/130; 13.84%), while either oligoxenous (40/143; 27.97%) or monoxenous species (100/269; 37.17%) had significantly higher prevalence (the latter two did not show significant differences between each other). Both mono- and oligoxenous flies hosted high diversities of individual Bartonella sequences (see Fig. 2.); however, the highest diversity of Bartonella strains was found in individual polyxenous species (Hpolyx = 1.831 vs. Hmonox = 1.779; Holigox = 1.556). Individual Nycteribiid fly species explained the distribution of Bartonella spp. only marginally, with only four species (N. vexata, Pe. conspicua, Pe. dufouri, Ph. biarticulatum) contributing significantly to the observed pattern (Table 2, and Fig. 3). Although these species define the probability of Bartonella occurrences in our sample, among these, high prevalence was established only in the case of N. vexata (18.18%) (Table 3). Other species with high prevalence were B. nattereri (9.53%) and N. pedicularia (9.09%). High prevalence was not associated with high diversity in case of Bartonella sequences, as different bat fly species hosted the highest number of individual Bartonella sequences (e.g., N. schmidlii (9), Pe. conspicua (7), and N. kolenatii (7), see also Fig. 3.).

The relationship between bat fly host specificity, bat fly species numbers, and individual Bartonella sequences, as well as genotypes recorded in bat flies in Central Europe

Unrooted tree representing phylogenetic relationships between bat fly species and Bartonella genotypes recorded
Association of Bartonella Strains with Bat Species
Bartonella-positive sequences were obtained from bat flies parasitizing 11 bat species (Miniopterus schreibersii, Myotis bechsteinii, My. blythii, My. capaccinii, My. daubentonii, My. myotis, My. nattereri, Rhinolophus blasii, R. euryale, R. ferrumequinum, and R. mehelyi), with only 3 bat species (Plecotus auritus, Pl. austriacus, and R. hipposideros) hosting only Bartonella-negative bat flies. The highest prevalence of Bartonella-positive bat flies were collected from R. euryale (5/7; 71.42%) and My. capaccinii (1/6; 33.33%), followed by Mi. schreibersii (78/240; 32.50%; see also Supplementary Table S1). Diversity of different Bartonella sequences was uneven among bat species, with the highest number of individual Bartonella sequences being recorded in bat flies hosted by Mi. schreibersii (11), followed by My. daubentonii (8) and My. blythii (6, see also Fig. 4). Bat species explained most of the variance found in Bartonella spp. distribution, with 6 individual species (Mi. schreibersii, My. blythii, My. capaccinii, My. daubentonii, My. myotis, and R. ferrumequinum) significantly contributing to the modeled distribution (Supplementary Table S3). Bat gender was another significant factor, with males holding more than twice as many Bartonella-positive bat flies as females (Table 4). Bat shelter also had an important contribution, as a significantly higher number of Bartonella-infected bat flies were collected on bats using underground shelters (Tables 4 and5), than from bats roosting either in buildings or in trees (no significant differences between the latter two groups).

Unrooted tree representing phylogenetic relationships between bat host species and Bartonella genotypes recorded
Genetic Heterogeneity and Sequence Clustering
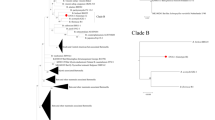
All sequences were confirmed genetically by amplification and sequencing of the gltA gene (sequences are listed as Supplementary Table S2). We identified 18 unique Bartonella sequences, which showed 95–100% similarity to Bartonella spp. sequences collected from bats (KF003129.1; KX300112) and bat flies (KT751145) (see also Table 3). As most of these sequences seem novel, they most likely represent undescribed Bartonella spp. associated with bats and their parasites, while they show similarity with pathogenic Bartonella spp., too (see Supplementary Table S4 and Supplementary Fig. S2 for a phylogenetic tree based on pairwise alignments with all the genotypes recorded together with similar Bartonella spp. genotypes from GenBank). These sequences clustered together into seven well-defined genotype groups (Genotypes 1 to 7, Figs. 4 and 5). Certain clusters showed wide geographical distribution (Genotypes 3 and 7, each with 10 different occurrences, found in both countries), while Genotypes 4 and 5 were restricted to southern Romania (Fig. 6). Most genotypes were shared by more than one bat fly species, while Genotypes 4 and 5 showed highly conservative distribution, being found only in parasites of the bats of the Rhinolophus genus (Fig. 7).

Unrooted tree representing phylogenetic relationships and the distribution of the seven recorded clusters of Bartonella sequences

Geographical distribution of the different Bartonella sequences clustered into the seven genospecies

Unrooted tree representing phylogenetic relationships between Bartonella genotypes recorded and bat-host families
Discussion
Our study detected a total of 18 unique Bartonella strains clustered in 7 genotypes, found in 9 out of the 10 bat fly species studied. The detection of Bartonella spp. DNA in 9/10 bat fly species (every third individual being positive) suggests that infection is highly prevalent in most bat fly populations, implying probable vectorial competence for these dipterans. Nycteribiidae bat flies were already suggested as vectors of different Bartonella spp. Several studies highlighted the importance of these flies as possible vectors for a series of Bartonella spp. [5, 25, 39, 40], while certain studies also suggest that the bat flies themselves are the reservoir hosts for these bacteria [23, 26]. We found high prevalence of Bartonella spp. in most bat fly species (overall prevalence 29.1%, range 6.2–66.0%), and also a wide range of geographical distribution (47.3%; 18 out of 39 collecting sites provided Bartonella-positive bat flies, with 100% prevalence for sites, where more than 10 flies were collected, n = 12 sites). This is in line with previous studies performed in tropical areas, where bat fly-Bartonella studies reported prevalences even higher than these figures (e.g., 87% for Cyclopodia dubia in Madagascar, [41]; 66% in Western Africa, [42]; up to 100% of Nycteribiidae in Costa Rica [43]) and fairly similar to data from temperate regions (38%, South Africa [44]). Our results are unique in terms of bat-associated Bartonella spp. sequence diversity detected in any temperate region. Previous studies in Europe recorded 12 sequences in France [45], four in Finland [29] while in UK only three sequences were detected [16].
The diversity of Bartonella genotypes found in the studied region is high, comparable only with tropical assemblages of bat-bat fly associations (Costa Rica, 34 strains in bats/bat flies [43]; Madagascar, 21 strains in bat flies [23]; or Western Africa, 39 strains in bats/bat flies [42]) and it was considered primarily a result of high vertebrate host- and ectoparasite diversity characterizing the tropical regions [23, 27, 40]. This is not the case for Central Europe, with its relatively small number of bat species (32, [31]) or Nycteribiidae bat flies (12 species, [3, 46].
The prevalence of Bartonella spp. bacteria in nycteribiid flies varied between the individual fly species and collection locations, with high prevalences noted especially in the case of N. kolenatii, N. schmidlii, and Pe. conspicua. Significantly higher prevalences were noted among oligoxenous and monoxenous bat fly species compared to polyxenous species. We hypothesize that this trend is maintained by the recurrent infection of individuals of mono- and oligoxenous bat flies feeding on a few infected individuals of their respective host species in contrast to polyxenous bat fly individuals which have access to more host individuals belonging to more bat host species due to their more promiscuous feeding regime [47]. Due to this habit, their feeding incidence on Bartonella-reservoir host individuals or species is likely more reduced. This is supported by our results of Bartonella spp. sequence diversity detected in individual polyxenous bat flies. As these fly species are promiscuous in host selection, they may acquire different Bartonella genotypes but since they do not always feed on the reservoir host (or they are not reservoir host themselves) they fail to maintain all these genotypes in the cycle for longer periods, causing the low prevalence detected in these flies. Individual Nycteribiidae fly species explained the distribution of Bartonella spp. only marginally, as only a few species contributed significantly to the observed pattern (Table 5). The reduced host species selection habit of mono- and oligoxenous parasites (Fig. 8) may favor the high prevalence of the detected Bartonella (these flies have to feed on the same infected host individuals of a selected number of species), while maintaining a reduced spectrum of Bartonella strain diversity in these host-vector cycles. Our study provides the first evidence of Bartonella spp. in these bat flies; thus, we lack any comparison with similar findings. We found no effect of bat fly gender on the distribution and diversity of Bartonella spp. DNA detection (although bat flies showed a highly skewed sex ratio, data not shown). We therefore hypothesize that this is caused by the large-scale movement of both genders of these flies among host individuals [48], thus providing equal chances of infection for flies of any gender.

Quantitative interaction web based on Bartonella genotype presence in samples of bat flies and their respective bat hosts. Links between nodes represent the sum of individual Bartonella genotype occurrences for a given bat and bat fly species couple
The occurrence of bat flies holding Bartonella spp. sequences show an uneven distribution among different bat species. While most bat species studied (11 out of 14) hosted infected bat flies, certain bat species were more prone to hold bacteria-positive flies. The highest prevalence of infected flies was recorded among bat species exclusively using caves (R. euryale, My. capaccini, and Mi. schreibersii). This is in line with their roosting ecology, as these species are spending the daylight hours in dense groups in traditional deep-cave roosts [31], thus providing easy access for flies to switch hosts among roosting bat individuals. This phenomenon is reassured by the low Bartonella spp. prevalence among flies hosted by bat species roosting in small groups (0 for both My. bechsteinii and My. natterreri) or individually inside caves (none in case of Plecotus spp., 1.6% for Rh. ferrumequinum). Thus, bat host species itself explained most of the variance found among Bartonella strain distribution (Table 3, Fig. 4). Bat gender is also an important factor regulating the distribution of Bartonella-positive bat flies. Male bats were hosting more than twice as many flies as females. While female bats spend most of their time in high cohesion social groups (both hibernating in winter, as well as in female-only reproductive colonies in summer), most males show a higher mobility during the year (Altringham and Senior 2005). As such, males of most tree roosting species switch roosts regularly in summer—some even at a daily basis [49, 50], and even cave or building-dwelling species are highly mobile both in summer [51] and in winter [52], seeking fertile females or visiting multiple lekking sites in swarming periods [53]. This high mobility presents wide opportunities for ectoparasites of these animals to access a large number of different host individuals. Bat gender was not an important factor in case of only one species, Mi. schreibersii, for which both genders roost together in colonies located solely in caves [31]. For this species, we found no difference among sexes, neither in prevalence nor in diversity of Bartonella-positive bat flies hosted. Bent-winged bats are long-distance migrants, with a number of stop-over sites along the migratory route [54]. This habit, coupled with their roosting preference of forming dense groups in high-number colonies (reaching up to thousand individuals all year long), may have favored the high diversity of Bartonella sequences (11 out of 18 molecularly identified) recorded in bat flies hosted by this bat species (see also Fig. 4).
In this study, we generated sequence data for 18 individual Bartonella strains from a number of nine bat fly species, which were collected from bats in Central Europe. These Bartonella sequences show 95–100% similarity to other Bartonella spp. reference sequences collected mostly from bats. However, none of the sequences from the present study are fully similar to any already known sequence collected from bat flies (the highest similarity is 99% with Bartonella spp. identified in bat flies from Madagascar, online Supplementary Table S5). All seven genotypes identified in our study are novel among bat fly-related Bartonella spp. and only show distant relationship to any previously described Bartonella species present in bat flies. This is in line with the wide diversity of Bartonella spp. identified from bats [21, 22, 24,25,26,27] or their parasites [28, 40,41,42,43,44]. The sequence characterization of a singular house-keeping gene (gltA) is not enough for establishing species boundaries among possible new Bartonella species; thus, we suggest that further characterization (culturing, multi-locus sequences) is necessary to verify whether the identified DNA sequences represent new Bartonella species or just variants of one or a few species [55]. Still, the identified genotypes seem generally distributed among a number of parasites (9 bat fly species) and bat species (11 bat species were hosting Bartonella-positive flies), with reduced specificity and a wide range of geographical records. While most sequences found show the highest similarity of bat-related Bartonella spp., several of the sequences identified are related to Bartonella spp. with known zoonotic potential (Bartonella washoensis, AF050108 or Bartonella koehlerae KX499329; 92% similarity for both). As most Bartonella-related diseases show a constantly emerging pattern [18, 20], with a number of known human cases caused by Bartonella spp., for which we lack any information regarding reservoir hosts or vectors [56], there is a need to establish the role of bats (and associated parasites) in the circulation of these bacteria. Moreover, the high diversity and prevalence detected in Central Europe suggest a wide and long-standing coevolution of these bacteria with Nycteribiidae and their insectivorous bat hosts, thus providing excellent study targets for close inspection of these host-vector-pathogen cycles (Fig. 8).
References
Tsang SM, Cirranello AL, Bates PJ, Simmons NB (2015) The roles of taxonomy and systematics in bat conservation. In: Voigt CC, Kingston T (eds) Bats in the Anthropocene: conservation of bats in a changing world. Springer International Publishing, pp 503–538
Teeling EC (2009) A molecular phylogeny for bats illuminates biogeography and the fossil record a molecular phylogeny for bats illuminates biogeography and the fossil record. Notes 307:580–584. https://doi.org/10.1126/science.1105113
Haelewaters D, Pfliegler WP, Szentiványi T, Földvári M, Sándor AD, Barti L, Camacho JJ, Gort G, Estók P, Hiller T, Dick CW, Pfister DH (2017) Parasites of parasites of bats: Laboulbeniales (Fungi: Ascomycota) on bat flies (Diptera: Nycteribiidae) in Central Europe. Parasit. Vectors 10:96. https://doi.org/10.1186/s13071-017-2022-y
Dick CW, Patterson BD (2006) Bat flies-obligate ectoparasites of bats. In: Morand S, Krasnov BR, RP (eds) Micromammals and macroparasites: from evolutionary ecology to management. Springer-Verlag, Tokyo Berlin Heidelberg New York, pp 179–194
Dick CW, Dittmar K (2014) Parasitic bat flies (Diptera: Streblidae and Nycteribiidae): host specificity and potential as vectors. In: Klimpel SMH (ed) Bats (Chiroptera) as vectors of diseases and parasites. Springer-Verlag, Berlin Heidelberg, pp 131–151
Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D, Bryden WL, Middleton D, Reid PA, McFarlane RA, Martin G, Tabor GM, Skerratt LF, Anderson DL, Crameri G, Quammen D, Jordan D, Freeman P, Wang LF, Epstein JH, Marsh GA, Kung NY, McCallum H (2015) Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B Biol. Sci. 282:20142124. https://doi.org/10.1098/rspb.2014.2124
Mühldorfer K (2013) Bats and bacterial pathogens: a review. Zoonoses Public Health 60:93–103. https://doi.org/10.1111/j.1863-2378.2012.01536.x
Hornok S, Szőke K, Kováts D, Estók P, Görföl T, Boldogh SA, Takács N, Kontschán J, Földvári G, Barti L, Corduneanu A, Sándor AD (2016) DNA of piroplasms of ruminants and dogs in ixodid bat ticks. PLoS One 11:e0167735. https://doi.org/10.1371/journal.pone.0167735
Corduneanu A, Hrazdilová K, Sándor AD, et al (2017) Babesia vesperuginis, a neglected piroplasmid: new host and geographical records, and phylogenetic relations. Parasit. Vectors 10:598. https://doi.org/10.1186/s13071-017-2424-x
Hornok S, Szőke K, Tu VT, Kontschán J, Takács N, Sándor AD, Halajian A, Földvári G, Estók P, Plantard O, Epis S, Görföl T (2017) Mitochondrial gene heterogeneity of the bat soft tick Argas vespertilionis (Ixodida: Argasidae) in the Palaearctic. Parasit. Vectors 10:109. https://doi.org/10.1186/s13071-017-2037-4
Gibson KE, Rikihisa Y, Zhang C, Martin C (2005) Neorickettsia risticii is vertically transmitted in the trematode Acanthatrium oregonense and horizontally transmitted to bats. Environ. Microbiol. 7:203–212. https://doi.org/10.1111/j.1462-2920.2004.00683.x
Evans NJ, Bown K, Timofte D, Simpson VR, Birtles RJ (2009) Fatal borreliosis in bat caused by relapsing fever spirochete, United Kingdom. Emerg. Infect. Dis. 15:1331–1333
Hosokawa T, Nikoh N, Koga R, Satô M, Tanahashi M, Meng XY, Fukatsu T (2012) Reductive genome evolution, host–symbiont co-speciation and uterine transmission of endosymbiotic bacteria in bat flies. ISME J 6:577–587. https://doi.org/10.1038/ismej.2011.125
Lei BR, Olival KJ (2014) Contrasting patterns in mammal-bacteria coevolution: Bartonella and Leptospira in bats and rodents. PLoS Negl. Trop. Dis. 8:e2738. https://doi.org/10.1371/journal.pntd.0002738
Hanson A (1970) Isolation of spirochaetes from primates and other mammalian species. Br J Vener Dis 46:303–306. https://doi.org/10.1136/sti.46.4.303
Concannon R, Wynn-Owen K, Simpson VR, Birtles RJ (2005) Molecular characterization of haemoparasites infecting bats (Microchiroptera) in Cornwall, UK. Parasitology 131:489–496. https://doi.org/10.1017/S0031182005008097
Segers FH, Kešnerová L, Kosoy M, Engel P (2017) Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J 11:1232–1244. https://doi.org/10.1038/ismej.2016.201
Boulouis HJ, Chang CC, Henn JB et al (2005) Factors associated with the rapid emergence of zoonotic Bartonella infections. Vet. Res. 36:383–410. https://doi.org/10.1051/vetres:2005009
Chomel BB, Boulouis HJ, Breitschwerdt EB, Kasten RW, Vayssier-Taussat M, Birtles RJ, Koehler JE, Dehio C (2009) Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet. Res. 40:29. https://doi.org/10.1051/vetres/2009011
Breitschwerdt EB (2014) Bartonellosis: one health perspectives for an emerging infectious disease. ILAR J. 55:46–58. https://doi.org/10.1093/ilar/ilu015
Kosoy M, Bai Y, Lynch T, Kuzmin IV, Niezgoda M, Franka R, Agwanda B, Breiman RF, Rupprecht CE (2010) Bartonella spp. in bats, Kenya. Emerg. Infect. Dis. 16:1875–1881. https://doi.org/10.3201/eid1612.100601
Bai Y, Hayman DTS, McKee CD, Kosoy MY (2015) Classification of Bartonella strains associated with straw-colored fruit bats (Eidolon helvum) across Africa using a multi-locus sequence typing platform. PLoS Negl. Trop. Dis. 9:e0003478. https://doi.org/10.1371/journal.pntd.0003478
Wilkinson DA, Duron O, Cordonin C, Gomard Y, Ramasindrazana B, Mavingui P, Goodman SM, Tortosa P (2016) The bacteriome of bat flies (Nycteribiidae) from the Malagasy region: a community shaped by host ecology, bacterial transmission mode, and host-vector specificity. Appl. Environ. Microbiol. 82:1778–1788. https://doi.org/10.1128/AEM.03505-15
Anh PH, Van Cuong N, Son NT et al (2015) Diversity of bartonella spp. in bats, southern Vietnam. Emerg. Infect. Dis. 21:1266–1267. https://doi.org/10.3201/eid2107.141760
Han HJ, Wen HL, Zhao L, Liu JW, Luo LM, Zhou CM, Qin XR, Zhu YL, Zheng XX, Yu XJ (2017) Novel Bartonella species in insectivorous bats, Northern China. PLoS One 12:e0167915. https://doi.org/10.1371/journal.pone.0167915
Olival KJ, Dittmar K, Bai Y, Rostal MK, Lei BR, Daszak P, Kosoy M (2015) Bartonella spp. in a Puerto Rican bat community. J. Wildl. Dis. 51:274–278. https://doi.org/10.7589/2014-04-113
McKee CD, Hayman DTS, Kosoy MY, Webb CT (2016) Phylogenetic and geographic patterns of bartonella host shifts among bat species. Infect Genet Evol 44:382–394. https://doi.org/10.1016/j.meegid.2016.07.033
Hornok S, Kovács R, Meli ML, Gönczi E, Hofmann-Lehmann R, Kontschán J, Gyuranecz M, Dán Á, Molnár V (2012) First detection of bartonellae in a broad range of bat ectoparasites. Vet. Microbiol. 159:541–543. https://doi.org/10.1016/j.vetmic.2012.04.003
Veikkolainen V, Vesterinen EJ, Lilley TM, Pulliainen AT (2014) Bats as reservoir hosts of human bacterial pathogen, Bartonella mayotimonensis. Emerg. Infect. Dis. 20:960–967. https://doi.org/10.3201/eid2006.130956
Haysom K, Dekker J, Russ J et al (2013) European bat population trends. A prototype biodiversity indicator, Technical. European Environment Agency, Bruxelles
Dietz C, von Helversen O, Nill D (2009) Bats of Britain, Europe and Northwest Africa. A & C Black, London
Theodor O (1967) An illustrated catalogue of the Rothschild collection of Nycteribiidae in the British Museum (Natural History), with keys and short descriptions for the identification of subfamilies, genera, species and subspecies. British Museum (Natural History) Publication 655, London
Theodor O, Moscona A (1954) On bat parasites in Palestine. I. Nycteribiidae, Streblidae, Hemiptera, Siphonaptera. Parasitology 44:157–245. https://doi.org/10.1017/S0031182000018862
Wielinga PR, Gaasenbeek C, Fonville M, de Boer A, de Vries A, Dimmers W, Akkerhuis op Jagers G, Schouls LM, Borgsteede F, van der Giessen JWB (2006) Longitudinal analysis of tick densities and Borrelia, Anaplasma, and Ehrlichia infections of Ixodes ricinus ticks in different habitat areas in the Netherlands. Appl. Environ. Microbiol. 72:7594–7601. https://doi.org/10.1128/AEM.01851-06
Norman a F, Regnery R, Jameson P et al (1995) Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J. Clin. Microbiol. 33:1797–1803
Dormann CF, Frund J, Bluthgen N, Gruber B (2009) Indices, graphs and null models: analyzing bipartite ecological networks. Open Ecol J 2:7–24. https://doi.org/10.2174/1874213000902010007
Core Team R (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Viena
Bates D, Maechler M, Bolker B, Walker S (2015) lm4: Linear mixded-effects models using Eigen and S4
Morse SF, Dick CW, Patterson BD, Dittmard K (2012) Some like it hot: evolution and ecology of novel endosymbionts in bat flies of cave-roosting bats (Hippoboscoidea, Nycterophiliinae). Appl. Environ. Microbiol. 78:8639–8649. https://doi.org/10.1128/AEM.02455-12
Morse SF, Olival KJ, Kosoy M et al (2012) Global distribution and genetic diversity of Bartonella in bat flies (Hippoboscoidea, Streblidae, Nycteribiidae). Infect Genet Evol 12:1717–1723. https://doi.org/10.1016/j.meegid.2012.06.009
Brook CE, Bai Y, Dobson AP, Osikowicz LM, Ranaivoson HC, Zhu Q, Kosoy MY, Dittmar K (2015) Bartonella spp. in fruit bats and blood-feeding ectoparasites in Madagascar. PLoS Negl. Trop. Dis. 10:e0003532. https://doi.org/10.1371/journal.pntd.0003532
Billeter S a, Hayman DTS, Peel aJ et al (2012) Bartonella species in bat flies (Diptera: Nycteribiidae) from western Africa. Parasitology 139:324–329. https://doi.org/10.1017/S0031182011002113
Judson SD, Frank HK, Hadly EA (2015) Bartonellae are prevalent and diverse in Costa Rican bats and bat flies. Zoonoses Public Health 62:609–617. https://doi.org/10.1111/zph.12188
Dietrich M, Tjale MA, Weyer J, Kearney T, Seamark ECJ, Nel LH, Monadjem A, Markotter W (2016) Diversity of bartonella and rickettsia spp. in bats and their blood-feeding Ectoparasites from South Africa and Swaziland. PLoS One 11:e0152077. https://doi.org/10.1371/journal.pone.0152077
Stuckey MJ, Boulouis HJ, Cliquet F, Picard-Meyer E, Servat A, Aréchiga-Ceballos N, Echevarría JE, Chomel BB (2017) Potentially zoonotic bartonella in bats from France and Spain. Emerg. Infect. Dis. 23:539–541. https://doi.org/10.3201/eid2303.160934
Szentiványi T, Estók P, Földvári M (2016) Checklist of host associations of European bat flies (Diptera: Nycteribiidae, Streblidae). Zootaxa 4205:101–126. https://doi.org/10.11646/zootaxa.4205.2.1
Gutiérrez R, Morick D, Cohen C, Hawlena H, Harrus S (2014) The effect of ecological and temporal factors on the composition of Bartonella infection in rodents and their fleas. ISME J 8:1598–1608. https://doi.org/10.1038/ismej.2014.22
Marshall AG (1982) Ecology of insects ectoparasitic on bats. In: Kunz TE (ed) Ecology of bats. Springer, US, Philadelphia, pp 369–401
Petit E, Mayer F (1999) Male dispersal in the noctule bat (Nyctalus noctula): where are the limits? Proc. Biol. Sci. 266:1717–1722. https://doi.org/10.1098/rspb.1999.0837
Encarnacao J a, Kierdorf U, Wolters V (2006) Seasonal variation in nocturnal activity of male Daubenton ’ s bats , Myotis daubentonii ( Chiroptera : Vespertilionidae). Folia Zool. 55:237–246
Moussy C, Hosken DJ, Mathews F, Smith GC, Aegerter JN, Bearhop S (2013) Migration and dispersal patterns of bats and their influence on genetic structure. Mamm Rev 43:183–195. https://doi.org/10.1111/j.1365-2907.2012.00218.x
Ramos Pereira MJ, Salgueiro P, Rodrigues L, Coelho MM, Palmeirim JM (2009) Population structure of a cave-dwelling bat, Miniopterus schreibersii: does it reflect history and social organization? J Hered 100:533–544. https://doi.org/10.1093/jhered/esp032
Altringham JD, Senior P, Ruckstuhl KE, Neuhaus P (2005) Social systems and ecology of bats. In: Ruckstuhl KE, Neuhaus P (eds) Sexual segregation in vertebrates. Cambridge University Press, Cambridge, pp 280–302
Rodrigues L, Palmeirim JM (2008) Migratory behaviour of the Schreiber’s bat: when, where and why do cave bats migrate in a Mediterranean region? J. Zool. 274:116–125. https://doi.org/10.1111/j.1469-7998.2007.00361.x
Gutiérrez R, Vayssier-Taussat M, Buffet J-P, Harrus S (2017) Guidelines for the isolation, molecular detection, and characterization of Bartonella species. Vector-Borne Zoonotic Dis 17:42–50. https://doi.org/10.1089/vbz.2016.1956
Kosoy M, Morway C, Sheff KW, Bai Y, Colborn J, Chalcraft L, Dowell SF, Peruski LF, Maloney SA, Baggett H, Sutthirattana S, Sidhirat A, Maruyama S, Kabeya H, Chomel BB, Kasten R, Popov V, Robinson J, Kruglov A, Petersen LR (2008) Bartonella tamiae sp. nov., a newly recognized pathogen isolated from three human patients from Thailand. J. Clin. Microbiol. 46:772–775. https://doi.org/10.1128/JCM.02120-07
Acknowledgements
Permission for bat capture was provided by the National Inspectorate for Environment, Nature and Water (Hungary), and the Underground Heritage Commission (Romania). Bat banding license numbers are 59/2003 (PE), 305/2015 (ADS), and TMF-493/3/2005 (TG). No live bat was harmed for this study. The authors thank C. Jére, I. Csősz, D. Bălășoiu, A. Telea, and A. Ionică for their contribution in the bat fly collection in Romania. We would like to express our thanks to Cristian Domșa for the help provided in the creation of the maps in Figs. 1 and 6. The survey was organized in the framework of the EurNegVec COST Action TD1303. This research was supported from the grant PN-II-RU-TE-2014-4-1389, the grant “In the light of evolution: theories and solutions” (GINOP-2.3.2-15-2016-00057), and the János Bolyai Research Scholarship of the Hungarian Academy of Science (to ADS and GF).
Author information
Authors and Affiliations
Contributions
ADS initiated the study, did part of the sample collection, and wrote the manuscript. LB, AC, SH, TG, PE, ZL, and DK contributed important samples to the study. MF identified all bat flies. AK, SSz, and HS performed the molecular and phylogenetic analyses. GF organized part of the sample collection and contributed to the study design and manuscript preparation. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Figure S1
Neighbor-joining phylogenetic tree based on the multiple alignment of the gltaA gene, including Bartonella sequences obtained in this study. Clusters were assigned based on visual inspection of the tree. (PDF 65 kb)
Supplementary Figure S2
Neighbor-joining phylogenetic tree based on the pairwise alignment of an approx. 360 bp long fragment of the gltaA gene, including representative Bartonella sequences obtained in this study (highlighted by green fonts and 5 character codes) and representative sequences of other Bartonella spp. retrieved from GenBank. Sequences in blue represent Bartonella spp. related to bats or bat flies, while sequences in black denote Bartonella spp. from other host species, Bartonella spp. with known pathogenicity are highlighted in bold. (PDF 31 kb)
Table S1
(XLSX 11 kb)
Table S2
(XLSX 35 kb)
Table S3
(XLSX 10 kb)
Table S4
(DOCX 15 kb)
Table S5
(XLSX 10 kb)
Rights and permissions
About this article

Cite this article
Sándor, A.D., Földvári, M., Krawczyk, A.I. et al. Eco-epidemiology of Novel Bartonella Genotypes from Parasitic Flies of Insectivorous Bats. Microb Ecol 76, 1076–1088 (2018). https://doi.org/10.1007/s00248-018-1195-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-018-1195-z