
Abstract
Microbial communities associated to the gut of insects are attracting an increasing interest, mainly because of their role in influencing several host life-traits. The characterization of the gut microbial community is pivotal for understanding insect ecology and, thus, to develop novel pest management strategies. The pine processionary moth, Thaumetopoea pytiocampa (Denis & Schiff.) (Lepidoptera: Thaumetopoeidae), is a severe defoliator of pine forests, able to feed on several pine species. In this work, we performed a metabarcoding analysis to investigate, for the first time, the diversity of the gut bacterial community of pine processionary larvae associated with three different host pine species (Pinus halepensis, Pinus nigra subsp. laricio, and Pinus pinaster). We found that the gut microbial community of T. pityocampa larvae collected on P. halapensis was different from that associated with larvae collected from P. nigra and P. pinaster. Moreover, the high presence of bacteria belonging to the genera Modestobacter, Delftia, and unidentified Methylobacteriaceae retrieved in larvae feeding on P. halapensis suggested that specific interactions can occur. Our results provide the evidence that different host plant differently impact on the microbiota diversity of T. pityocampa larvae, contributing to the general knowledge of this pest with information that could be useful in shaping the next generation of pest control strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Most of the microorganisms are known to have a cosmopolitan distribution, and interacting with other organisms can structure and modulate several biotic interactions [1, 2]. Recently, the microbial communities associated with insects attracted increasing interest, mainly because of their ecological and economical importance. Indeed, microorganisms possess metabolic properties that are often absent in insects so, in this way, they can provide traits which enable insects to overcome plant defenses [3]. This is particularly clear in herbivorous insects because plant tissues, having a wide range of indigestible and toxic compounds, do not represent a promising food for them. Therefore, insects evolved a series of strategies to overcome plant defenses [4], including the association with bacterial and fungal symbionts to exploit different hosts [5, 6]. The interaction between insects and microorganisms can be extended at different levels of the trophic ladder and can also comprise indirect interactions mediated by plants [7].
The ecological interactions between gut bacteria and insects have a great impact on various host life-traits contributing, for example, to food digestion, production of essential vitamins, and to counteract potentially harmful microbes [8]. Insect gut microbiota demonstrates also a certain degree of plasticity, with the ability to adapt rapidly to changes in the insect diet, even with modifications in the microbial population structure [9]. This plasticity, on the other hand, can be useful for insects to exploit different food sources, providing the basis for the development of host-associated differentiation (HAD), which is an adaptive ecological radiation that could explain the high species diversity observed in insects [10,11,12,13]. Thus, the characterization of the gut microbial community is essential for a comprehensive understanding of the biology and ecology of the host insect and, potentially, can lead to the development of novel pest management strategies. This appears particularly attractive not only for insect pest of agricultural interest but also for insects that are harmful to forests, where pest control programs are limited and/or difficult to realize [14].
The pine processionary moth, Thaumetopoea pytiocampa (Denis & Schiff.) (Lepidoptera: Thaumetopoeidae), is one the most devastating defoliator insect of pine forests in Southern Europe and North Africa. This moth is univoltine, with polyphagous larvae feeding on Pinus spp., Cedrus spp., and Larix spp. [15]. Development requires the passage through five larval instars during the autumn and winter. Larvae are strongly gregarious; during winter, they group inside silky nests which provide protection from low temperatures. Mature larvae have urticating setae, which can cause severe health problems in warm-blooded animals, including humans, such as dermatitis, conjunctivitis, and anaphylaxis. Throughout the Italian territory, T. pityocampa affects the growth of several pine species, resulting the most destructive pest in pine plantations [16, 17]. The host plant and the forest type play an essential role on the susceptibility to processionary attacks, due to an active process of host detection and selection by adult female moths prior to oviposition, and/or differential survival of eggs or immature larvae [16]. Pine species were largely used in Italy for reforestation programs started at the beginning of last century, favoring the spread of the pine processionary caterpillar [17]. Insect attacks are still a key problem in the management of pine-woods for many areas, which seem to be linked to the forest structure, and in particular to the species of pine, the age of the trees, and the stand density [17]. This appears particularly true in Southern Italy, where the Laricio pine (Pinus nigra subsp. laricio Poir. (Maire)) has been widely used in reforestation, although no forest management or forestry operations have been carried out to maintain the improved landscape, resulting extremely vulnerable to abiotic and biotic agents [18].
Therefore, the comprehension of the relationship between plant-insect-microorganisms is essential to understand the ecology of this system. The output of these interactions could be predictable for monophagous species, which survival is linked to a single plant species. Thus, it is questionable how the gut microbial community reacts when an insect can exploit different plant species as food source. The answer to this question can be essential in planning the future generation of eco-friendly pest management practices in forest systems, like the attempts currently under development in other biological systems [19,20,21]. In this work, we aimed to characterize the gut bacterial community of pine processionary larvae associated with three different host pine species (Pinus halepensis Miller, P. nigra subsp. laricio, and Pinus pinaster Aiton) in order to get information about the microbial community associated to this pest, and whether it changes according to different host plants.
Materials and Methods
Insect Samplings and Storage
Larvae of T. pityocampa at the fourth instar were collected in January 2016 at Aspromonte National Park (Reggio Calabria, Italy, 38° 9′ 32.76″ N–15° 55′ 14.16″ E), directly from nests collected from three different Pinus species. We chose to study the fourth larval instar, since it represents a mature larva, which has been feeding on the same host species for several weeks; thus, it is supposed to have been developed an adapted gut microbiota. Host plant species were selected on the basis of the different susceptibility to the processionary moth [15]: the Aleppo pine (P. halepensis), the Maritime pine (P. pinaster), and the Laricio pine (P. nigra subsp. laricio). For each host, we selected 3 plants outdistanced about 20 m among them and from which we collected 5 larvae (15 specimens per host plant). Immediately after collection, larvae were kept at 4 °C until transportation to the laboratory and then placed at −80 °C. Dead insects were then sterilized superficially with a solution of sodium hypochlorite (21.6 g/L) for 5 min, washed once in ethanol (70% v/v) for 2 min and finally three times in sterile distilled water for 10 s. Dead larvae were then placed in phosphate buffer solution to excise the complete gut with sterile forceps. Samples were then stored at −80 °C until DNA extraction.
DNA Extraction and Library Preparation
Gut tissues were crushed in the extraction buffer (10 mM Tris, 100 mM NaCl, 10 mM EDTA, 0.5% SDS) with the aid of a bead mill homogenizer (three 1 mm∅ stainless steel beads per 1.5-ml tube, milled for 5 min at 30 Hz), and the mixture was treated with proteinase K (5Prime GmbH, Germany) following the producer’s instructions [22]. Total DNA was extracted using the MoBio PowerSoil Kit (Mo Bio Laboratories, Inc., Carlsbad, CA, USA) following the manufacturer’s protocol [23]. DNA concentration and quality were assessed by means of a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., USA).
The bacterial community of 15 specimens from each sampled host plant was characterized using a metabarcoding approach targeting the bacterial 16S rRNA. To avoid the co-amplification of eukaryotic small subunit ribosomal RNA (SSU) rDNA, a nested PCR strategy was used. For the external PCR amplification, we used the primer Eub8F [24] and the broad coverage primer 984yR [25]. Then, the V4 hyper-variable region of 16S rRNA region was amplified using the 16S rRNA primers 515f/806rB developed by Caporaso et al. [26], following the procedure described by Caporaso et al. [27]. Each amplification was carried out in triplicate, including a negative control with nuclease-free water instead of DNA. Amplification success was checked by electrophoresis on 1.5% agarose gel stained with GelRed (Biotium Inc., Fremont, CA, USA). PCR products from the same sample were then pooled together to reduce stochasticity errors [28] and cleaned using Agencourt AMPure XP kit (Beckman Coulter, Brea, CA, USA). A further short-run PCR was performed to integrate Illumina i7 and i5 indexes following the producer’s protocol (Nextera XT, Illumina, San Diego, CA, USA), and amplicons were purified again as explained above. Libraries were then quantified with the Invitrogen Qubit™ dsDNA HS Assay Kit (Invitrogen, Carlsbad, CA, USA), normalized to a concentration of 10 ng/μl using nuclease-free water, pooled together, and sequenced with the Illumina MiSeq sequencer, using the MiSeq Reagent Kit v3 600-cycles chemistry (Illumina, San Diego, CA, USA) following the producer’s protocol.
Data Analysis
Demultiplexed forward and reverse reads were merged using PEAR 0.9.1 algorithm applying default parameters [29]. Data handling was carried out using QIIME 1.9 [30], quality filtering reads, binning OTUs using open-reference OTU-picking through UCLUST algorithm, discarding chimeric sequences discovered with USEARCH 6.1, and assigning taxonomy querying towards GreenGenes database using the BLAST method. Singletons and OTUs retrieved in less than five samples were discarded from the downstream analyses. Samples with number of sequences <9000 were discarded, i.e., seven samples (four from P. pinaster and three from P. nigra subsp. laricio) were not used in the analyses.
R statistical software [31], plugged with the packages vegan, phyloseq, and DESeq2 [32,32,34] was used to calculate the species accumulation curves (SACs) and diversity indices for each sample. Multivariate analyses were performed comparing samples by host plant by a PERMANOVA analysis, supported by Tukey’s MCT and visualizations by DPCoA (double principal coordinate analysis). DESeq2 was used to filter the OTUs differentially present according to host plant, and the GLM procedure with Tukey’s MCT was performed to test their abundance on larvae collected from different plants.
Results
Analyses yielded a total of 1,396,775 reads after quality check, with an average of 36,757 sequences per sample. In order to be processed, reads were clustered by a similarity of 0.97, ending up to a total of 1007 OTUs. As demonstrated by SACs (Fig. S1), the sequencing depth per sample was deep enough to uncover most of the bacterial diversity in insects’ guts. Also, the Shannon’s diversity index resulted homogeneous among the insects sampled on the on the tested Pinus spp. (Fig. S2). An overall analysis indicated that most of the sequences from the bacterial community associated with T. pityocampa developing on different Pinus spp. belonged to Proteobacteria (61.03 ± 2.68%), followed by Actinobacteria (19.1 ± 3.23%), Bacteroidetes (14.69 ± 1.57%), and Firmicutes (2.53 ± 0.45%). Proteobacteria resulted the highly dominant and diverse phylum, represented by all four classes: α-Proteobacteria (11.4 ± 0.78%), β-Proteobacteria (7.62 ± 1.22%), γ-Proteobacteria (41.82 ± 2.99%), and δ-proteobacteria (0.2 ± 0.04%). γ-Proteobacteria, the most abundant lineage within Proteobacteria, was represented by Moraxellaceae (33.06 ± 2.9%), followed by Enterobacteriaceae (3.88 ± 1.25%) and Pseudomonadaceae (3.15 ± 0.5%).
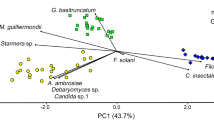
Comparing the microbial community of insects collected on the different plants, using PERMANOVA multivariate nonparametric test, differences among the three plant species were highlighted (F 3, 38 = 6.02; P < 0.001). The Tukey’s MCT evidenced differences between P. halepensis and the other two host plants (P < 0.01), but not between P. nigra subsp. laricio and P. pinaster (P > 0.05). This result is also depicted in the DPCoA graph (Fig. 1), which shows that larvae collected on P. halepensis clustered separately from those collected on the other Pinus species.

Double principal coordinate analysis (DPCoA) performed on the whole microbial community associated to larvae of Thaumetopoea pityocampa feeding on different host plant species (Pinus halepensis, Pinus nigra subsp. laricio, and Pinus pinaster). Ellipses are calculated at the 95% confidence interval
A deeper analysis of the dataset allowed to identify 41 OTUs, which were differently abundant among insects associated with the three different plant species (P < 0.01). These were further filtered taking into account only taxa that represented at least 1% of the microbiota in larvae feeding on different host plant, allowing to detect 28 OTUs representing seven taxa with likely a major role in T. pityocampa ability to exploit different host plants (Table 1, Fig. 2). Larvae collected on P. halepensis were characterized by a higher amount of Actinobacteria (40.43 ± 4.03%) compared with insects collected on P. nigra subsp. laricio (4.94 ± 0.53%) and P. pinaster (5.45 ± 0.6%). More specifically, this effect was associated with the differential abundance of 8 OTUs associated with the genus Modestobacter (38.4 ± 4.05%), which resulted more abundant in P. halepensis than P. nigra subsp. laricio and P. pinaster (Table 1). Differently, the phylum Proteobacteria was less abundant in larvae feeding on P. halepensis (46.68 ± 3.2%) than those collected on P. nigra subsp. laricio (73.38 ± 3.68%) or P. pinaster (67.15 ± 2.57%). However, a different pattern arises when analyzing this lineage more in detail. Specifically, 2 OTUs belonging to Methylobacteriaceae, 1 OTU associated with unidentified Pseudomonadaceae, and 12 OTUs associated with the genus Delftia were more abundant in larvae collected from P. halepensis, whereas 1 OTU associated with Pseudomonas and 3 OTUs with Acinetobacter were more abundant on larvae collected from both P. pinaster and P. nigra subsp. laricio (Table 1). Finally, 2 OTUs associated with unidentified Comamonadaceae were more abundant only in larvae feeding on P. nigra subsp. laricio.

Boxplot representing the differential abundance of key bacterial taxa in the microbial community associated to larvae of Thaumetopoea pityocampa feeding on different host plants (Pinus halepensis, Pinus nigra subsp. laricio, and Pinus pinaster)
Discussions
We analyzed for the first time the diversity of the gut bacterial community of T. pityocampa, which composition resulted considerably affected by the three host pine species tested in this study. Our results fit in a wider framework, which sees a recurrent interplay between host plant and herbivore, influencing to each other. This kind of interaction has been previously observed, for example, in the oligophagous aphid Aphis citricidus (Kirkaldy) [35] and in the pea aphid Acyrthosiphon pisum (Harris) [36]. It has been argued therefore that the main drivers that shape insect gut microbial community are diet, life stage, and environment [9]. Symbionts have been investigated for the effects on their host, directly by mediating interactions with other species, or indirectly by impacting the host genetic diversity, with effects that could be extended at the community level [37]. These symbionts could be vertically transferred from one generation to the next, horizontally among individuals of the same species, but can also be acquired directly from the environment [38]. Moreover, symbionts can influence insect response to plant defenses, provide protection from natural enemies, and influence the reproductive system [37]. These are just few examples of the plasticity of insect-microorganism relationship. On the other hand, this association can be so strict that experimental exchanges of gut microbial communities in herbivorous insects lead to a decrease in performance on host plant [39]. Although several studies investigated bacterial communities associated to insects, current knowledge on the fungal microbiota is still quite restricted and the available studies highlight unexpected ecological interactions between insects and fungi [22, 40, 41]. The characterization of insect microbial communities, together with information on any host-associated variation in microbiota composition, is essential for a comprehensive understanding of insect ecology as well as for the development of novel pest management strategies. For example, as outlined by Crotti et al. [42], microbes can be used to enhance SIT programs, to counteract the spread of insect-borne pathogens and their vectors and to protect beneficial insects. To develop such techniques, baseline knowledge on the biology of the target species, and the characterization of its associated microorganisms, needs to be accomplished [43], including both life stage and host-associated microbiota.
In this study, we found that the population of gut symbionts of T. pityocampa larvae associated with P. halapensis was different from the symbionts associated with larvae that fed on P. nigra subsp. laricio and P. pinaster. Pinus halapensis is mainly found in Aspromonte pinewoods within mixed forest at low altitude (approx. 350–500 m a.s.l.), whereas P. nigra subsp. laricio and P. pinaster are both mainly located at higher altitude (>700 m a.s.l.) [44]. It has been reported that insects’ microbiota is geographically stable considering the same host plant [45,45,47]; therefore, it is unlikely that in our study, the location played a primary role. Further investigations may help to shed some light on this aspect. On the other hand, as the gut of lepidopteran larvae has a relatively simple morphology [48] lacking specific structures for harboring microbial symbionts, it is not surprising that the gut microbiota can be affected by diet [49], as we experienced in our study. The exploiting of different microbial communities on different diets could also contribute to explain the different susceptibility of the Pinus species to the attack by T. pityocampa. Indeed, EPPO [15] provided an exhaustive list of hosts of pine processionary moth, ranking them for their susceptibility to this pest. In this case, P. nigra subsp. laricio is considered more susceptible than P. halepensis and P. pinaster. However, studies conducted in the Mediterranean basin suggested a different order of pine species preference, which seems to vary accordingly to the geographical regions or types of experiments [16]. The different host species susceptibility could play an important role in shaping T. pityocampa microbial community, as Macchioni et al. [50] reported that the main constituent in the essential oils extracted from the needles of P. halepensis was myrcene, whereas the one obtained from the needles of P. nigra, and P. pinaster, showed higher amounts of α-pinene. Since it is acknowledged that microbial symbionts can help insects to counteract plant defense mechanisms [51], the lower overall susceptibility of P. halepensis to T. pytiocampa can explain the different composition of pine processionary gut microbial community, when compared to specimens collected on the other host plants. For this instance, our results suggest that Actinobacteria and Methylobacteriaceae, more abundant in insects feeding on P. halepensis, may play an important role.
Proteobacteria was the dominant phylum in pine processionary gut microbiome, which has been extensively found to be associated with other insects [9, 52]. Mohr et al. [53] identified the genus Delftia associated with some species of bees. Furthermore, Acinetobacter johnsonii was identified to be associated with mosquitoes [54] and Dendroctonus valens LeConte [55]. Methylobacteriaceae have been discovered to be associated with Bactrocera tau (Walker), Helicoverpa armigera (Hüb.), and lycaenid butterflies [56,56,58]. Members of Comamonadaceae and Pseudomonaceae were identified in our study and already reported to be associated to Anopheles gambiae Giles [59]. Beyond their identification, the role of these taxa continues to be undisclosed although, as observed by Russel et al. [60] in different species of ants, bacteria belonging to Burkholderiales, Pseudomonadales, and Rhizobiales are widely associated with herbivore habits. Furthermore, we identified a high presence of Actinobacteria associated with T. pityocampa. This bacterial lineage has been previously reported for other insects, for example pyrrhocorid bugs, where it looks to act as essential nutritional symbionts [61]. In general, Actinobacteria associated with insects provide protection against detrimental microorganisms [62]. Interestingly, among Actinobacteria, reads associated with the genus Modestobacter were more abundant in T. pityocampa larvae associated with P. halapensis than in larvae collected from the other two Pinus species. This result is of particular interest from an ecological point of view, since it suggests a specific association between Modestobacter and T. pityocampa larvae feeding on P. halapensis. Little is known about genus Modestobacter. This genus belongs to the actinobacterial family Geodermatophilaceae [63], which contains species that are typical inhabitants of exposed surfaces such as monuments and natural stones [64] or surface soils [65] and arid soils [66]. Pinus halapensis success in colonizing the less hospitable rocks prefers warmer calcareous areas and is more tolerant to drought compared to P. nigra subsp. laricio and P. pinaster [67]. Thus, we do not exclude that the presence of member belonging to the genus Modestobacter in the T. pityocampa gut might have a role in the pine processionary moth adaptation to this species of Pinus. Unfortunately, for many species, the relationship between the insect and its microbiota remain undefined. Recent studies reported the association of genus Modestobacter with the microbial community of the sweet potato whitefly, Bemisia tabaci (Gennadius) B biotype [68], and with the herbivore beetle Cryptocephalus marginellus Olivier [69]. Otherwise, we cannot exclude that Modestobacter might be acquired by T. pityocampa from P. halapensis during the evolutionary process of adaptation and might contribute to the establishment of a permanent association [70]. Further studies dealing with the genetic diversity of pine processionary moths associated with different host plants can be pivotal to disentangle this aspect.
This study provides the first information about the microbial diversity associated with T. pytiocampa, showing that the microbiota of larvae collected on P. halepensis was different from those sampled on P. nigra subsp. laricio and P. pinaster. Our results support the idea that the different host plant differently impact on the diversity of the microbiome associated with larvae of pine processionary moth. It is not excluded that heritable symbionts vertically transmitted may have a role in the mechanism of T. pytiocampa adaptation to different hosts and habitats, but further studies are needed to investigate this aspect. Bacteria associated with insects can confer their host partner with traits allowing them to differentially exploit distinct host plant species, defining the species host range [71, 72]. These results could be successfully extended in other geographical areas, to test the stability of the microbial community along latitudinal and altitudinal gradients, and to other plant species, to better understand which pattern drives the diversification of gut microbial community. These information could be essential in shaping the future generation of pest management strategies.
Data Accessibility
Raw sequence reads from Illumina sequencing are available on NCBI SRA database under the Project Accession ID: PRJNA379119.
References
Prosser JI (2015) Dispersing misconceptions and identifying opportunities for the use of ‘omics’ in soil microbial ecology Nat Rev Micro 13:439–446. doi:10.1038/nrmicro3468
Whitman WB, Coleman DC, Wiebe WJ (1998) Prokaryotes: the unseen majority Proc. Natl. Acad. Sci. U. S. A. 95:6578–6583
Dillon R, Dillon V (2004) The gut bacteria of insects: nonpathogenic interactions Annual Reviews in Entomology 49:71–92
Goldman-Huertas B, Mitchell RF, Lapoint RT, Faucher CP, Hildebrand JG, Whiteman NK (2015) Evolution of herbivory in Drosophilidae linked to loss of behaviors, antennal responses, odorant receptors, and ancestral diet Proc. Natl. Acad. Sci. U. S. A. 112:3026–3031. doi:10.1073/pnas.1424656112
Frago E, Dicke M, Godfray HCJ (2012) Insect symbionts as hidden players in insect–plant interactions Trends Ecol. Evol. 27:705–711. doi:10.1016/j.tree.2012.08.013
Brady CM, White JA (2013) Cowpea aphid (Aphis craccivora) associated with different host plants has different facultative endosymbionts Ecol Entomol 38:433–437. doi:10.1111/een.12020
Biere A, Bennett AE (2013) Three-way interactions between plants, microbes and insects Funct. Ecol. 27:567–573. doi:10.1111/1365-2435.12100
Feldhaar H (2011) Bacterial symbionts as mediators of ecologically important traits of insect hosts Ecol Entomol 36:533–543. doi:10.1111/j.1365-2311.2011.01318.x
Colman DR, Toolson EC, Takacs-Vesbach CD (2012) Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21:5124–5137. doi:10.1111/j.1365-294X.2012.05752.x
Abrahamson WG, Weis AE (1997) Evolutionary ecology across three trophic levels: goldenrods, gallmakers, and natural enemies. Princeton University Press
Berlocher SH, Feder JL (2002) Sympatric speciation in phytophagous insects: moving beyond controversy? Annu. Rev. Entomol. 47:773–815
Drès M, Mallet J (2002) Host races in plant–feeding insects and their importance in sympatric speciation Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 357:471–492
Jaenike J (1981) Criteria for ascertaining the existence of host races Am. Nat. 117:830–834
Wojciechowska M, Stepnowski P, Gołębiowski M (2016) The use of insecticides to control insect pests Invertebr. Surviv. J. 13:210–220
EPPO (2004) Thaumetopoea pityocampa- PM7/37 Bulletin OEPP/EPPO Bulletin 34:295–298
Jactel H, Barbaro L, Battisti A, Bosc A, Branco M, Brockerhoff E, Castagneyrol B, Dulaurent A-M, Hódar JA, Jacquet J-S (2015) Insect–tree interactions in Thaumetopoea pityocampa. Processionary moths and climate change: an update. Springer, pp. 265–310
Masutti L, Battisti A (1990) Thaumetopoea pityocampa (Den. & Schiff.) in Italy bionomics and perspectives of integrated control J. Appl. Entomol. 110:229–234
Avolio S, Bernardini V, Clerici E, Tomaiuolo M (2012) Management guidelines for calabrian pine reforestations carried out in southern Italy in the 1950s-70s J Life Sci 6:1050
Campolo O, Malacrinò A, Grande SB, Chiera E, Palmeri V (2015) Efficacy of selected insecticides for the control of the california red scale in southern Italy Acta Hort 1065:1149–1156
Campolo O, Palmeri V, Malacrinò A, Laudani F, Castracani C, Mori A, Grasso DA (2015) Interaction between ants and the Mediterranean fruit fly: new insights for biological control Biol. Control 90:120–127
Malacrinò A, Campolo O, Laudani F, Palmeri V (2016) Fumigant and repellent activity of limonene enantiomers against Tribolium confusum du Val Neotropical Entomol 45:597–603
Malacrinò A, Schena L, Campolo O, Laudani F, Palmeri V (2015) Molecular analysis of the fungal microbiome associated with the olive fruit fly Bactrocera oleae Fungal Ecol. 18:67–74. doi:10.1016/j.funeco.2015.08.006
Rubin BE, Sanders JG, Hampton-Marcell J, Owens SM, Gilbert JA, Moreau CS (2014) DNA extraction protocols cause differences in 16S rRNA amplicon sequencing efficiency but not in community profile composition or structure MicrobiologyOpen 3:910–921
Weisburg WG, Barns SM, Pelletier DA, Lane DJ (1991) 16S ribosomal DNA amplification for phylogenetic study J. Bacteriol. 173:697–703
Wang Y, Qian P-Y (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies PLoS One 4:e7401
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms ISME J 6:1621–1624
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci 108(Supplement 1):4516–4522
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W, Bolchacova E, Voigt K, Crous PW (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi Proc. Natl. Acad. Sci. U. S. A. 109:6241–6246
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina Paired-End reAd mergeR Bioinformatics 30:614–620
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data Nat. Methods 7:335–336
R Core Team (2013) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Online: http://www.r-project.org/.
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2 Genome Biol. 15:550
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217.
Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin P, O’Hara R, Simpson G, Solymos P, Stevens M, Wagner H (2013) Package “vegan”: community ecology. R package version: 2.0–7
Guidolin AS, Cônsoli FL (2017) Symbiont diversity of Aphis (Toxoptera) citricidus (Hemiptera: Aphididae) as influenced by host plants Microb. Ecol. 73:201–210
Ferrari J, West JA, Via S, Godfray HCJ (2012) Population genetic structure and secondary symbionts in host-associated populations of the pea aphid complex Evolution 66:375–390
Ferrari J, Vavre F (2011) Bacterial symbionts in insects or the story of communities affecting communities Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 366:1389–1400
Kikuchi Y, Hosokawa T, Fukatsu T (2007) Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation Appl. Environ. Microbiol. 73:4308–4316. doi:10.1128/aem.00067-07
Hosokawa T, Kikuchi Y, Shimada M, Fukatsu T (2007) Obligate symbiont involved in pest status of host insect Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 274:1979–1984. doi:10.1098/rspb.2007.0620
Malacrinò A, Schena L, Campolo O, Laudani F, Mosca S, Giunti G, Strano CP, Palmeri V (2017) A metabarcoding survey on the fungal microbiota associated to the olive fruit fly Microb. Ecol. 73:677–684. doi:10.1007/s00248-016-0864-z
Malacrinò A, Rassati D, Schena L, Mehzabin R, Battisti A, Palmeri V (2017) Fungal communities associated with bark and ambrosia beetles trapped at international harbours Fungal Ecol. 28:44–52. doi:10.1016/j.funeco.2017.04.007
Crotti E, Balloi A, Hamdi C, Sansonno L, Marzorati M, Gonella E, Favia G, Cherif A, Bandi C, Alma A, Daffonchio D (2012) Microbial symbionts: a resource for the management of insect-related problems Microb. Biotechnol. 5:307–317. doi:10.1111/j.1751-7915.2011.00312.x
Andongma AA, Wan L, Dong Y-C, Li P, Desneux N, White JA, Niu C-Y (2015) Pyrosequencing reveals a shift in symbiotic bacteria populations across life stages of Bactrocera dorsalis Sci Rep 5:9470. doi:10.1038/srep09470
Palmeri V, Pulvirenti A, Zappalà L (2005) La processionaria dei pini nei boschi della dorsale appenninica della Calabria Forest@-Journal of Silviculture and Forest Ecology 2:345
Berasategui A, Axelsson K, Nordlander G, Schmidt A, Borg-Karlson A-K, Gershenzon J, Terenius O, Kaltenpoth M (2016) The gut microbiota of the pine weevil is similar across Europe and resembles that of other conifer-feeding beetles Mol. Ecol. 25:4014–4031. doi:10.1111/mec.13702
Gayatri Priya N, Ojha A, Kajla MK, Raj A, Rajagopal R (2012) Host plant induced variation in gut bacteria of Helicoverpa armigera PLoS One 7:e30768. doi:10.1371/journal.pone.0030768
Sudakaran S, Salem H, Kost C, Kaltenpoth M (2012) Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae) Mol. Ecol. 21:6134–6151. doi:10.1111/mec.12027
Engel P, Moran NA (2013) The gut microbiota of insects–diversity in structure and function FEMS Microbiol. Rev. 37:699–735
Broderick NA, Raffa KF, Goodman RM, Handelsman J (2004) Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods Appl. Environ. Microbiol. 70:293–300
Macchioni F, Cioni P, Flamini G, Morelli I, Maccioni S, Ansaldi M (2003) Chemical composition of essential oils from needles, branches and cones of Pinus pinea, P. halepensis, P. pinaster and P. nigra from central ltaly Flavour Fragrance J 18:139–143
Hansen AK, Moran NA (2014) The impact of microbial symbionts on host plant utilization by herbivorous insects Mol. Ecol. 23:1473–1496. doi:10.1111/mec.12421
Yun J-H, Roh SW, Whon TW, Jung M-J, Kim M-S, Park D-S, Yoon C, Nam Y-D, Kim Y-J, Choi J-H, Kim J-Y, Shin N-R, Kim S-H, Lee W-J, Bae J-W (2014) Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host Appl. Environ. Microbiol. 80:5254–5264. doi:10.1128/aem.01226-14
Mohr KI, Tebbe CC (2006) Diversity and phylotype consistency of bacteria in the guts of three bee species (Apoidea) at an oilseed rape field Environ. Microbiol. 8:258–272. doi:10.1111/j.1462-2920.2005.00893.x
Pidiyar VJ, Jangid K, Patole MS, Shouche YS (2004) Studies on cultured and uncultured microbiota of wild Culex quinquefasciatus mosquito midgut based on 16s ribosomal RNA gene analysis. Am J Trop Med Hyg 70:597–603. doi:10.4269/ajtmh.2004.70.597
Morales-Jiménez J, Zúñiga G, Villa-Tanaca L, Hernández-Rodríguez C (2009) Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae) Microb. Ecol. 58:879–891. doi:10.1007/s00248-009-9548-2
Prabhakar CS, Sood P, Kanwar SS, Sharma PN, Kumar A, Mehta PK (2013) Isolation and characterization of gut bacteria of fruit fly, Bactrocera tau (Walker) Phytoparasitica 41:193–201. doi:10.1007/s12600-012-0278-5
Whitaker MRL, Salzman S, Sanders J, Kaltenpoth M, Pierce NE (2016) Microbial communities of lycaenid butterflies do not correlate with larval diet Front. Microbiol. 7:1920. doi:10.3389/fmicb.2016.01920
Paramasiva I, Shouche Y, Kulkarni GJ, Krishnayya PV, Akbar SM, Sharma HC (2014) Diversity in gut microflora of Helicoverpa armigera populations from different regions in relation to biological activity of Bacillus thuringiensis δ-endotoxin Cry1Ac Arch. Insect Biochem. Physiol. 87:201–213. doi:10.1002/arch.21190
Wang Y, Gilbreath III TM, Kukutla P, Yan G, Xu J (2011) Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya PLoS One 6:e24767. doi:10.1371/journal.pone.0024767
Russell JA, Moreau CS, Goldman-Huertas B, Fujiwara M, Lohman DJ, Pierce NE (2009) Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants Proc. Natl. Acad. Sci. U. S. A. 106:21236–21241. doi:10.1073/pnas.0907926106
Salem H, Kreutzer E, Sudakaran S, Kaltenpoth M (2013) Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae) Environ. Microbiol. 15:1956–1968. doi:10.1111/1462-2920.12001
Kaltenpoth M (2009) Actinobacteria as mutualists: general healthcare for insects? Trends Microbiol. 17:529–535. doi:10.1016/j.tim.2009.09.006
Normand P (2006) Geodermatophilaceae fam. nov., a formal description Int. J. Syst. Evol. Microbiol. 56:2277–2278
Urzì C, Salamone P, Schumann P, Rohde M, Stackebrandt E (2004) Blastococcus saxobsidens sp. nov., and emended descriptions of the genus Blastococcus Ahrens and Moll 1970 and Blastococcus aggregatus Ahrens and Moll 1970 Int. J. Syst. Evol. Microbiol. 54:253–259
Luedemann GM (1968) Geodermatophilus, a new genus of the Dermatophilaceae (Actinomycetales) J. Bacteriol. 96:1848–1858
Reddy GS, Potrafka RM, Garcia-Pichel F (2007) Modestobacter versicolor sp. nov., an actinobacterium from biological soil crusts that produces melanins under oligotrophy, with emended descriptions of the genus Modestobacter and Modestobacter multiseptatus Mevs et al. 2000 Int. J. Syst. Evol. Microbiol. 57:2014–2020
Fuentes D, Disante KB, Valdecantos A, Cortina J, Vallejo VR (2007) Response of Pinus halepensis Mill. seedlings to biosolids enriched with Cu, Ni and Zn in three Mediterranean forest soils Environ. Pollut. 145:316–323
Indiragandhi P, Yoon C, Yang JO, Cho S, Sa TM, Kim GH (2010) Microbial communities in the developmental stages of B and Q biotypes of sweetpotato whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae) J. Korean Soc. Appl. Biol. Chem. 53:605–617. doi:10.3839/jksabc.2010.093
Montagna M, Gómez-Zurita J, Giorgi A, Epis S, Lozzia G, Bandi C (2015) Metamicrobiomics in herbivore beetles of the genus Cryptocephalus (Chrysomelidae): toward the understanding of ecological determinants in insect symbiosis Insect Sci 22:340–352
Hosokawa T, Ishii Y, Nikoh N, Fujie M, Satoh N, Fukatsu T (2016) Obligate bacterial mutualists evolving from environmental bacteria in natural insect populations Nat Microbiol 1:15011. doi:10.1038/nmicrobiol.2015.11
Medina RF, Nachappa P, Tamborindeguy C (2011) Differences in bacterial diversity of host-associated populations of Phylloxera notabilis Pergande (Hemiptera: Phylloxeridae) in pecan and water hickory J. Evol. Biol. 24:761–771. doi:10.1111/j.1420-9101.2010.02215.x
Shin SC, Kim S-H, You H, Kim B, Kim AC, Lee K-A, Yoon J-H, Ryu J-H, Lee W-J (2011) Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling Science 334:670–674. doi:10.1126/science.1212782
Acknowledgments
Analyses were carried out using instruments acquired with the support of PON SAF@MED (PON a3_00016) and PON PON03PE_00090_1-2-3 (PON Ricerca e competitività 2007–2013).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Strano, C.P., Malacrinò, A., Campolo, O. et al. Influence of Host Plant on Thaumetopoea pityocampa Gut Bacterial Community. Microb Ecol 75, 487–494 (2018). https://doi.org/10.1007/s00248-017-1019-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-017-1019-6