
Abstract
The cutaneous microbiota plays a significant role in the biology of their vertebrate hosts, and its composition is known to be influenced both by host and environment, with captive conditions often altering alpha diversity. Here, we compare the cutaneous bacterial communities of 61 amphibians (both wild and captive) from Hiroshima, Japan, using high-throughput amplicon sequencing of a segment of the 16S rRNA gene. The majority of these samples came from a captive breeding facility at Hiroshima University where specimens from six species are maintained under highly standardized conditions for several generations. This allowed to identify host effects on the bacterial communities under near identical environmental conditions in captivity. We found the structure of the cutaneous bacterial community significantly differing between wild and captive individuals of newts, Cynops pyrrhogaster, with a higher alpha diversity found in the wild individuals. Community structure also showed distinct patterns when comparing different species of amphibians kept under highly similar conditions, revealing an intrinsic host effect. Bacterial communities of dorsal vs. ventral skin surfaces did not significantly differ in most species, but a trend of higher alpha diversity on the ventral surface was found in Oriental fire-bellied toads, Bombina orientalis. This study confirms the cutaneous microbiota of amphibians as a highly dynamic system influenced by a complex interplay of numerous factors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The integument of animals is the first level of protection against invasive pathogens from the environment, but represents more than a physical barrier. In vertebrates, a chemical defense is provided by the active compounds secreted by skin glands in combination with metabolites produced by microbes inhabiting the skin [1, 2]. The role of symbiotic skin bacteria in host immunity has been intensively studied [3, 4]. These microorganisms are known to contribute to and modulate the immune responses in the host [5, 6]. After its initial establishment, the microbiota of the skin is highly dynamic and throughout the entire life of the host, microbes are constantly acquired and lost [7].
In amphibians, the skin provides an incomplete physical barrier and active compounds within the mucosal environment play an important role in defense against pathogens [8]. Many of these compounds, such as alkaloids and anti-microbial peptides, originate from the host and are known to interact with and inhibit pathogens [9–11]. Other compounds originate from skin-associated bacteria, and are also known to play a role in suppressing pathogens themselves and to interact synergistically with host-produced compounds [12–15].
As in humans and other vertebrates, the composition of the cutaneous bacterial communities on amphibians is dynamic, and influenced by multiple factors, including host species and environment [16–22]. Additionally, the skin is a heterogeneous structure, with differences in texture and density of glands among body regions [23], which can influence the composition of skin-associated bacteria [24].
To better understand how cutaneous microbial communities influence the health of their amphibian hosts, it is essential to characterize the composition and specificity of these communities. In the present study, we assessed variation of the skin bacterial communities of six species of amphibians (Bombina orientalis, Bufo japonicus, Cynops pyrrhogaster, Echinotriton andersoni, Odorrana splendida, and Rana japonica) from one of the largest amphibian breeding facilities worldwide. In this center, located in Hiroshima University in Japan, different species of amphibians are kept under highly standardized conditions for several generations, providing ideal and novel conditions for a comparative assessment of host effects on the microbial community structure because it removes the confounding effects of inhabiting different environments. In addition, this study provides information on the composition of the skin microbial community of Asian amphibians which are still poorly studied. We analyzed samples from this collection and from wild-caught specimens from Hiroshima University campus, to explore to which degree cutaneous bacterial communities differ in composition and diversity (1) between wild and captive conspecific amphibians, (2) between species kept under highly similar conditions in captivity, and (3) between dorsal and ventral body surface in captive amphibians.
Material and Methods
Sampling
During the first week of March 2015, 61 adult amphibians belonging to six species housed at the breeding facility of the Institute for Amphibian Biology, Hiroshima University, were sampled for skin bacterial communities: Oriental fire-bellied toads, Bombina orientalis; Japanese common toads, Bufo japonicus; Japanese fire-bellied newts, Cynops pyrrhogaster; Anderson’s crocodile newts, Echinotriton andersoni; Amami Ishikawa’s frogs, Odorrana splendida; and Japanese brown frogs, Rana japonica. This captive collection was initiated with wild individuals in 1994 (for Rana japonica) and currently is composed of wild caught to F2 generations for the amphibian species used here. For this study, we sampled 7–14 individuals per species, with typically only 1–2 specimens from a single cage to reduce pseudoreplicates (Table 1; exceptions were B. orientalis with three individuals from two cages each and E. andersoni with five individuals from one cage). Specimens in this collection are maintained for the purpose of other research and conservation breeding projects, and sufficient large sample sizes from independent cages could not be obtained in all cases for a single sex. Because exploratory analyses of our dataset did not yield any difference in composition or diversity of the bacterial communities between the sexes, we combined data from male and female individuals for analysis. Individuals from all species (with exception of breeding groups of E. andersoni) are kept in similar environmental conditions, which consist of plastic terraria (61 × 40 × 15 cm) with a gravel substrate of 2–3-cm depth. There is continuous flow of water from the upper side and a drainage pipe at 1-cm level which keeps the environment wet, thus preventing drying of their skin. Water of all cages comes from the same source. E. andersoni individuals can also be kept in the same gravel-substrate containers, but for breeding they are kept in breeding cage (90 × 90 × 50 cm) with a wet muddy bottom (a mixture of 70:30 v/v leaves and soil used as floor cover).
Additionally, nine C. pyrrhogaster from a shallow pond and adjacent ditch (ca. 10–20-cm water depth; approximately 50 × 20 m) within the Hiroshima University campus were sampled. Wild individuals were captured by dip netting or manually with gloved hands, sampled on the ventral surface as described below and released quickly, assuring the shortest handling time possible. All sampled individuals were adults.
Sampling technique was non-invasive and consisted of rinsing the sampling surface of the amphibian with 50 ml of autoclaved filtered doubled distilled water to remove transient bacteria and then rubbing the cleaned surface ten times with a synthetic cotton swab (MW113; Medical Wire & Equipment, Corsham, UK). Clean nitrile gloves were used to hold and swab each individual. Swabs were individually placed in sterile 1.5 ml centrifuge tubes and stored at −20 °C until DNA extraction.
Sample Processing
DNA was extracted from the swabs with MoBio Power Soil htp-96 extraction kits (MoBio Laboratories, Carlsbad, CA, USA) with the minor adjustments outlined in Kueneman et al. [20] and double centrifugation time to account for the available rotor speed. The V4 region [25] of the bacterial 16S rRNA gene was amplified and sequenced using the dual-index approach of Kozich et al. [26].
All PCRs were performed on a T1 thermal cycler (Biometra) in duplicate. Individual reactions (12.5 μl) were composed by 0.15 μl of Phusion Hot Start II DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA), 0.25 μl of each forward (515 F: 5′-GTGCCAGCMGCCGCGGTAA-3′) and reverse primer (806R: 5′-GGACTACHVGGGTWTCTAAT-3′) (10 μM), 0.25 μl of dNTP, 2.5 μl of buffer (as supplied with the polymerase; Thermo Scientific), 8.1 μl of H2O, and 1 μl of template DNA. The protocol consisted on an initial denaturation step at 98 °C for 1 min, then amplification during 30 cycles at 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 30 s and a final extension of 5 min at 72 °C. After amplification, the two duplicate PCR products were combined (25 μl per sample) and visualized on 1 % agarose gels.
Samples were pooled for sequencing by collecting approximately equal amounts of each samples’ PCR product and gel purified with the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA). The purified pool concentration was determined with the Broad-Range dsDNA kit on a Qbit 2.0 and the pooled samples were submitted for Illumina Miseq sequencing using paired-end 2 × 250 v2 chemistry at the Genome Center of the Helmholtz Center for Infection Biology in Braunschweig, Germany.
The sequences of the amplicon libraries were deposited in the NCBI short read database (Bioproject PRJNA320971).
Sequence Processing
All sequences were processed with Quantitative Insights Into Microbial Ecology (MacQIIME v1.9.1) unless otherwise stated [27]. Following the default settings of the Fastq-join, the raw forward and reverse 16S rRNA amplicons from each sample were paired [28, 29]. After the initial pairing, sequences underwent quality filtering to remove low-quality sequences. Sequences were additionally filtered by read length (250–253 base pairs; usegalaxy.org). The identification of chimeras was performed using usearch61 de novo based detection on a per sample basis (http://drive5.com/usearch/usearch_docs.html; [30]). Chimeric sequences, i.e., sequences originating from PCR artifacts and composed of partial sequences of different bacteria, were removed before picking operational taxonomic units (OTUs). Using an open reference OTU-picking strategy, sequences were clustered into OTUs at 97 % similarity (http://qiime.org/tutorials/open_reference_illumina_processing.html; [31]). First, the sequences were matched to the SILVA 119 database (24 July 2014; https://www.arb-silva.de) and the remaining sequences were de novo clustered at a 97 % similarity. The UCLUST [32] algorithm was used in both the reference-based and de novo clustering steps. The reference sequence for each OTU was chosen based on which sequence was most abundant for a given OTU. The taxonomy of the representative sequences was determined by aligning them with PyNAST [33] and assigning taxonomy using the RDP classifier [34] with the SILVA 119 taxonomy. A phylogenetic tree was built with FastTree [35] under default settings. Based on Bokulich et al. [36], a cut-off was made removing all OTUs with less than 0.005 % of the total reads, leaving 4,812,392 reads in the dataset. To standardize the number of reads across samples for our main analyses, all samples were rarefied to 1000 sequences per sample, and samples with fewer than 1000 sequences were removed.
Statistical Analyses
The overall data was divided in three sets to allow the following comparisons: (1) Comparisons between wild and captive individuals. These comparisons were restricted to C. pyrrhogaster and targeted the ventral skin bacterial community of nine captive and nine wild individuals. (2) Comparisons between species. These comparisons were made among five amphibian species that were kept under similar conditions for several generations. This analysis included the ventral skin samples from ten B. orientalis, seven B. japonicus, nine C. pyrrhogaster, ten O. splendida, and fourteen R. japonica. (3) Comparison between body regions (i.e., ventral versus dorsal). These comparisons used the same individuals as the species comparison plus 11 E. andersoni.
Within each dataset, the data was divided into subgroups according to species and/or sample type (e.g., ventral swabs of B. orientalis, dorsal swabs of B. orientalis). The core bacterial community at the OUT level shared by at least 80 % of the samples in each sub-group was determined and all of the different cores merged. The datasets were then filtered for these merged core OTUs and the information on their proportion in each taxon visualized in bar plots.
Species richness was assessed using the Chao1 and OTU indices, and phylogenetic diversity was assessed using Faith’s PD index. These indices were calculated within QIIME for all subgroups of the data using the entire datasets. These metrics were compared for each comparison group in R [37] version 3.3.2 with linear regression models and ANOVAs after confirming statistical normality of the dataset. A dissimilarity matrix among the samples was calculated for the samples within each comparison group outlined above using the Bray-Curtis and the unweighted UniFrac metric within QIIME [38]. These matrices were further analyzed in Primer 7 by non-metric multidimensional scaling (nMDS) and by performing a PERMANOVA to statistically analyze the three comparisons outlined above. If main effects were significant, subsequent pair-wise PERMANOVAs were completed. LEfSe analyses were performed on the Galaxy web-based interface (http://huttenhower.sph.harvard.edu/galaxy/) using standard parameters except the alpha value for the factorial Kruskal-Wallis test among classes (alpha = 0.01).
Core OTUs that were shared by 80 % of the samples per category were included in the comparisons represented by Venn diagrams, generated manually in CorelDraw X3 (Corel Corp.).
Results
Effect of Captivity Conditions
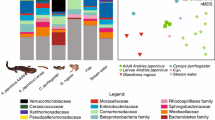
The cutaneous bacterial communities from captive C. pyrrhogaster were compared to those from a wild population at the same location. The 80 % core bacterial community in captivity was dominated by Xantomonadaceae (23.9 %) and Pseudomonaceae (18.9 %), while in the wild it was dominated by Comamonadaceae (44.9 %) and Verrucomicrobiaceae (19.2 %) (Fig. 1a, supplementary material, Fig. S1). Wild individuals had more diverse communities (Chao1 index; t test; P value = 0.044), and a tendency towards a greater phylogenetic diversity (Faith’s PD index; t test; P value = 0.081), but similar OTU richness (t test: P value = 0.625) compared to captive individuals. The two groups analyzed were significantly different from each other in beta diversity (PERMANOVA: P value = 0.001) (Fig. 1b). The 80 % core of the communities from wild and captive individuals consisted of 35 and 23 OTUs, respectively, and 14 of these OTUs were shared between the two categories (Fig. 1c, supplementary material, Table S1). A LEfSe analysis revealed 107 taxa distinguishing the skin bacterial communities of wild and captive specimens. From these taxa, 69 and 38 occurred in higher proportions in wild and captive individuals, respectively. The phyla Acidobacteria, Actinobacteria, Fusobacteria, Tenericutes, and Verrucomicrobia were detected as more abundant for the wild individuals, while Spirochaetae was more abundant in the captive individuals. At the genus level Acidothermus, Micrococcus, Marmoricola, Cloacibacterium, Sphingobacterium, Anaerotruncus, Bradyrhizobium, Ralstonia, Vogesella, Pseudomonas, and Opitutus were significantly more abundant in the wild individuals, while Ferruginibacter, Mucilaginibacter, Sphingomonas, Massilia, Thermomonas, and Spirochaeta were significantly more abundant in captive individuals. Additionally, at the OTU level, 29 and 19 OTUs were more abundant in wild and captive individuals, respectively (supplementary material, Fig. S2).

a Composition of the 80 % core skin bacterial communities of wild and captive Cynops pyrrhogaster at the genus level. Most abundant genera are identified in the figure (full legend in supplementary material, Fig. S1). b Plot from a two-dimensional nMDS analysis, based on unweighted UniFrac distances between skin bacterial communities of wild and captive samples of C. pyrrhogaster. c Venn diagram showing numbers of 80 % core OTUs unique to and shared between wild and captive C. pyrrhogaster
Host Species Effect
Five amphibian species (B. orientalis, B. japonicus, C. pyrrhogaster, O. splendida, and R. japonica) kept under the same conditions at the breeding facility of Hiroshima University were analyzed. The 80 % core skin bacterial community was largely dominated by Flavobacteriaceae, followed by Moraxellaceae and Pseudomonaceae in B. japonicus, O. splendida, and R. japonica. In B. orientalis and C. pyrrhogaster, Pseudomonaceae was the most abundant family followed by unclassified Micrococcales and Flavobacteriaceae in the first species and Xanthomonadaceae and Comamonadaceae in the second (Fig. 2). ANOVA revealed differences in alpha diversity between the analyzed groups (P value < 0.001). Pair-wise tests between skin bacterial communities from the different species confirmed consistent significant differences for the number of OTUs, Chao1, and Faith’s PD indexes for the pairs: B. japonicus–R. japonica and O. splendida–R. japonica, being R. japonica less OTU rich and alpha diverse in both cases (Table 2, Fig. 2). The bacterial communities clustered according to species and were all significantly different from each other in beta diversity (Fig. 3a, supplementary material, Table S2). At the 80 % core OTU level, B. orientalis had 41 OTUs, B. japonicus had 43 OTUs, C. pyrrhogaster had 23 OTUs, O. splendida had 51 OTUs, and R. japonica had 32 OTUs. From these, six OTUs were shared among all species (Fig. 3b, supplementary material, Table S3). A LEfSe analysis revealed 16 taxa which distinguished the skin bacterial communities of the various species. Ten of these were associated with B. orientalis, one with B. japonicus, one with C. pyrrhogaster, two with O. splendida, and two with R. japonica (supplementary material, Fig. S3).

Composition of the 80 % core skin bacterial communities of the analyzed species both dorsally and ventrally at the genus level. Most abundant bacterial genera are identified in the figure (full legend in supplementary material, Fig. S1). Unclass: group not classified bellow the order level. Other: group not classified bellow family level

a Plots from a two-dimensional nMDS analysis (unweighted UniFrac) representing the species-related differences in the composition of the skin bacterial communities. b Venn diagram showing number of 80 % core OTUs shared between the six analyzed species
Body Region Effect
The dorsal and ventral surfaces of six amphibian species (B. orientalis, B. japonicus, C. pyrrhogaster, E. andersoni, O. splendida, and R. japonica) were compared. For all species, the 80 % core skin bacterial community of the dorsal and ventral surfaces were dominated by the same families and had similar abundances. Exceptions were found in B. orientalis and C. pyrrhogaster. On the ventral surface of B. orientalis, the abundance of Flavobacteriaceae was larger, while Pseudomonaceae and unclassified Micrococcales exhibited greater relative abundance dorsally. On the dorsum of C. pyrrhogaster, the abundance of Xanthomonadaceae was reduced when compared to the ventral surface, with Pseudomonaceae and Flavobacteriaceae being the most abundant families (Fig. 2). Differences in beta diversity were consistent across individuals as indicated by the clustering of bacterial communities according to body region in the nMDS plot (Fig. 4a, Table 3). For all species, the majority of core OTUs were shared between ventral and dorsal surfaces (Fig. 4b, supplementary material, Tables S4 to S9). With the exception of B. orientalis, no species showed differences in OTU richness or alpha diversity indices between dorsal and ventral cutaneous bacterial communities. In this species, all the metrics showed significant differences between the surfaces (Table 3), with larger values characterizing the ventral surface.

a Plots from two-dimensional nMDS analyses (unweighted UniFrac) representing the body region-related differences in the composition of the skin bacterial communities. b Venn diagrams showing number of 80 % core OTUs shared between the dorsal and ventral body surfaces for the six analyzed species
Discussion
Our study adds to the evidence characterizing the cutaneous bacterial communities of amphibians as being influenced by captivity, host species, and body region (e.g., [17, 20]). Bacterial communities of the skin of captive and wild Japanese amphibians were dominated by Gamma-Proteobacteria, Bacteroidetes and in some amphibian taxa Actinobacteria, which is in agreement with findings from amphibians in North America [17, 20], Central America [39, 40], and Europe [41]. Our study, herein, provides evidence that captive environment, body region, and host identity influence the community composition.
To explore how the captive environment at the Hiroshima amphibian breeding facility affects cutaneous bacterial communities, we focused on the comparison between wild and captive C. pyrrhogaster. Some of the captive newts were direct descendants from the sampled wild population which is in close proximity (∼300 m) to the breeding facility. Yet, the skin bacterial community between both groups was strongly divergent. We also found a decreased alpha diversity of cutaneous bacteria in captive newts, which agrees with previous studies that consistently point towards a loss of alpha diversity in captivity [18, 19, 21, 24, 42]. This reduction in diversity may lead to a higher susceptibility of the hosts to diseases [12] and therefore needs to be considered for ex situ management of threatened amphibians, especially in projects that have as an objective of re-introducing individuals into the wild.
The breeding facility of the Institute of Amphibian Biology of Hiroshima University provides an exceptional setting for testing how the host factors influence skin-associated skin bacterial communities. Differences in cutaneous bacterial communities between species have regularly been found in syntopic wild amphibians (e.g., [17, 20]). However, in natural settings, confounding factors usually cannot be excluded, e.g., a treefrog and an aquatic frog might differ in skin microbiota either due to intrinsic host effects or due to their different microhabitat usage. In the Hiroshima breeding facility, most amphibians are kept under standardized conditions, i.e., in cages of the same size, same substrate, same water supply, same food, and largely at the same temperature. All specimens included in the present study had been kept under homogeneous conditions for several years. The skin bacterial communities of these individuals would be expected to be identical if the environmental factors were the sole driving forces acting on it, while species-specific communities would suggest that host factors are shaping the community composition. The identity and proportion of core genera were relatively constant between host species, suggesting the role of environmental factors. Chryseobacterium was the most abundant bacterial genus, except for the fire-bellied toad B. orientalis and the newt C. pyrrhogaster on which Pseudomonas and a taxon representing an unidentified Xanthomonadaceae genus were respectively most abundant. Still, we found significant differences in bacterial community structure between all five amphibian species compared (B. orientalis, B. japonicus, C. pyrrhogaster, O. splendida, and R. japonica). Despite some overlap in the nMDS plot, all individuals roughly clustered by species, reflecting the existence of intrinsic host effects in structuring the cutaneous microbial community. In the sixth species in our study, E. andersoni, specimens were kept under heterogeneous conditions, and it was therefore not included in the inter-species comparisons. In this species, individuals seem to cluster by tank (data not presented due to low sample size), reflecting once again the strong influence of environmental factors on the cutaneous microbiota [21].
Comparisons among bacterial communities of dorsal and ventral skin surfaces yielded no significant differences in five amphibian species (B. japonicus, C. pyrrhogaster, E. andersoni, O. splendida, and R. japonica) (Figs. 2 and 4, Table 3). However, in Oriental fire-bellied toads (B. orientalis), the ventral side had a higher species richness and phylogenetic diversity. This result is consistent with the analyses of Bataille et al. [24] in the same species, and could be related both to the different roughness of the dorsal and ventral sides, and to the toxin secretion (such as anti-microbial peptides) from the dorsal skin [43].
The composition and diversity of the bacterial communities of amphibians are determined by a multitude of factors, and disentangling these is a challenging task. Although amphibians at Hiroshima University are mainly kept for conservation of biological and genetic studies, the settings at this breeding facility allowed for an isolated testing of these factors, keeping other variables more constant than in natural settings. Simultaneous analyses of newts from the breeding center and their population of origin demonstrated how captive conditions changes and especially reduces the diversity of bacterial communities, probably due to overall reduced environmental complexity in the breeding tanks. Comparison among captive amphibians kept under highly similar conditions over a long time provided strong support for host-specific characteristics influencing cutaneous bacteria, and the differences between dorsal and ventral communities in only Oriental fire-bellied toads points to an effect of dorsal poison-secreting glands on the microbiota.
Less than a third of the 80 % core OTUs were shared between the groups in the wild-captive (shared OTUs = 32 %) and species (shared OTUs = 30 %) comparisons, while approximately two thirds where shared between body regions (shared OTUs = 62 %). Overall, as a testable hypothesis for future studies, we posit that external environmental factors (including those related to captive conditions) are as important as intrinsic host-related effects on influencing the composition and diversity of amphibian cutaneous microbiotas. Different parts of the amphibian body, on the contrary, usually bear similar cutaneous microbiotas, except in species where skin on different body parts produces secretions with distinct chemical properties.
Numerous other factors influencing amphibian-associated bacterial communities remain to be explored, such as temperature effects [44], the effect of pathogens on the community [40], and the emergence of opportunistic pathogenic bacteria in diseased and lesioned specimens. Most available studies, including the one presented here, focus on comparisons of the more abundant members of the bacterial communities, but future work on the rare OTUs is warranted as these can have fundamental functions for the host [45]. In general, the importance of the skin bacterial community to host health is well established [46]. An improved causal understanding of its composition and variation, and development of methods to maintain and manipulate it, can become fundamental for conservation management of captive and wild amphibian populations.
Change history
28 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00248-021-01693-z
References
Clarke BT (1997) The natural history of amphibian skin secretions, their normal function and potential medical applications. Biol Rev 72:365–379
Rosenthal M, Goldberg D, Aiello A, Larson E, Foxman B (2011) Skin microbiota: microbial community structure and its potential association with health and disease. Infect Genet Evol 11:839–848
Grice EA, Segre JA (2011) The skin microbiome. Nat Rev Microbiol 9:244–253
Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W et al (2012) Compartmentalized control of skin immunity by resident commensals. Science 337:1115–1119
Belden LK, Harris RN (2007) Infectious diseases in wildlife: the community ecology context. Front Ecol Environ 5:533–539
Reid G, Younes JA, Van Der Mei HC, Gloor GB, Knight R, Busscher HJ (2011) Microbiota restoration: natural and supplemented recovery of human microbial communities. Nat Rev Microbiol 9:27–38
Fierer N, Ferrenberg S, Flores GE, Gonzalez A, Kueneman J, Legg T et al (2012) From animalcules to an ecosystem: application of ecological concepts of the human microbiome. Ann Rev Ecol Evol S 43:137–155
Woodhams DC, Brandt H, Baumgartner S, Kielgast J, Küpfer E, Tobler U et al (2014) Interacting symbionts and immunity in the amphibian skin mucosome predict disease risk and probiotic effectiveness. PLoS One 9, e96375
Daly JW, Myers CW, Whittaker N (1987) Further classification of skin alkaloids from Neotropical poison frogs (Dendrobatidae), with a general survey of toxic/noxious substances in the amphibia. Toxicon 25:1023–1095
Woodhams DC, Ardipradja K, Alford RA, Marantelli G, Reinert LK, Rollins‐Smith LA (2007) Resistance to chytridiomycosis varies among amphibian species and is correlated with skin peptide defenses. Anim Conserv 10:409–417
Rollins-Smith LA (2009) The role of amphibian antimicrobial peptides in protection of amphibians from pathogens linked to global amphibian declines. BBA-Biomembranes 1788:1593–1599
Becker MH, Harris RN (2010) Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS One 5, e10957
Myers JM, Ramsey JP, Blackman AL, Nichols AE, Minbiole KP, Harris RN (2012) Synergistic inhibition of the lethal fungal pathogen Batrachochytrium dendrobatidis: the combined effect of symbiotic bacterial metabolites and antimicrobial peptides of the frog Rana muscosa. J Chem Ecol 38:958–965
Bletz MC, Loudon AH, Becker MH, Bell SC, Woodhams DC, Minbiole KP, Harris RN (2013) Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol Lett 16:807–820
Walke JB, Becker MH, Loftus SC, House LL, Teotonio TL, Minbiole KP, Belden LK (2015) Community structure and function of amphibian skin microbes: an experiment with bullfrogs exposed to a chytrid fungus. PLoS One 10, e0139848
Harris RN, Lauer A, Simon MA, Banning JL, Alford RA (2008) Addition of antifungal skin bacteria to salamanders ameliorates the effects of chytridiomycosis. Dis Aquat Org 83:11
McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL (2012) Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6:588–596
Antwis RE, Haworth RL, Engelmoer DJ, Ogilvy V, Fidgett AL, Preziosi RF (2014) Ex situ diet influences the bacterial community associated with the skin of red-eyed tree frogs (Agalychnis callidryas). PLoS One 9, e85563
Becker MH, Richards-Zawacki CL, Gratwicke B, Belden LK (2014) The effect of captivity on the cutaneous bacterial community of the critically endangered Panamanian golden frog (Atelopus zeteki). Biol Conserv 176:199–206
Kueneman JG, Parfrey L, Woodhams DC, Archer HM, Knight R, McKenzie VJ (2014) The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol 23:1238–1250
Loudon AH, Woodhams DC, Parfrey LW, Archer H, Knight R, McKenzie V, Harris RN (2014) Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J 8:830–840
Walke JB, Becker MH, Loftus SC, House LL, Cormier G, Jensen RV, Belden LK (2014) Amphibian skin may select for rare environmental microbes. ISME J 8:2207–2217
Toledo RD, Jared C (1995) Cutaneous granular glands and amphibian venoms. Comp Biochem Phys A 111:1–29
Bataille A, Lee-Cruz L, Tripathi B, Kim H, Waldman B (2016) Microbiome variation across amphibian skin regions: implications for chytridiomycosis mitigation efforts. Microb Ecol 71:221–232
Brosius J, Dull TJ, Sleeter DD, Noller HF (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J Mol Biol 148:107–127
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD (2013) Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Aronesty E (2011) ea–utils: command–line tools for processing biological sequencing data. http://code.google.com/p/ea–utils. Accessed 1 Oct 2015
Aronesty E (2013) TOBioiJ: comparison of sequencing utility programs. Open Bioinforma J 7:8
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM (2014) Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. Peer J 2, e545
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Price MN, Dehal PS, Arkin AP (2010) FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality–filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59
R Development Core Team (2011) R: a language and environment for statistical computing. Vienna: the R Foundation for Statistical Computing. ISBN: 3-900051-07-0. Available online at http://www.R-project.org/
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Belden LK, Hughey MC, Rebollar EA, Umile TP, Loftus SC, Burzynski EA, Minbiole KP, House LL, Jensen RV, Becker MH, Walke JB, Medina D, Ibáñez R, Harris RN (2015) Panamanian frog species host unique skin bacterial communities. Front Microbiol 6:1171
Rebollar EA, Hughey MC, Medina D, Harris RN, Ibáñez R, Belden LK (2016) Skin bacterial diversity of Panamanian frogs is associated with host susceptibility and presence of Batrachochytrium dendrobatidis. ISME J. doi:10.1038/ismej.2015.234
Vences M, Dohrmann AB, Künzel S, Granzow S, Baines JF, Tebbe CC (2015) Composition and variation of the skin microbiota in sympatric species of European newts (Salamandridae). Amphibia-Reptilia 36:5–12
Kohl KD, Skopec MM, Dearing MD (2014) Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv Physiol 2, cou009
Gibson BW, Tang DZ, Mandrell R, Kelly M, Spindel ER (1991) Bombinin-like peptides with antimicrobial activity from skin secretions of the Asian toad, Bombina orientalis. J Biol Chem 266:23103–23111
Kohl KD, Yahn J (2016) Effects of environmental temperature on the gut microbial communities of tadpoles. Environ Microbiol, in press
Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA (2014) Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5, e01371-14
Sanford JA, Gallo RL (2013) Functions of the skin microbiota in health and disease. Semin Immunol 25:370–377
Acknowledgments
We are grateful to Meike Kondermann for their help in the lab and to Christoph Tebbe for helpful advice. We express our appreciation to the Board of Education of Kagoshima prefecture for allowing us to use live crocodile newts and Amami Ishikawa’s frogs protected by law. We thank the strain maintenance team of the Institute for Amphibian Biology for providing captive Japanese fire-bellied newts. This work was supported by a grant of the Deutsche Forschungsgemeinschaft (VE247/9-1) and by a guest researcher fellowship of Hiroshima University to MV.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Fig. S1
Full legend for the taxa bar plots. Percentages of each taxa represented for each subset of the data. (PDF 52.9 kb)
Supplementary Fig. S2
Results from LEfSe analysis showing taxa that significantly differ in abundance between wild and captive C. pyrrhogaster. Green taxa significantly characterize the wild community and blue taxa the captive community. (PDF 45.2 kb)
Supplementary Fig. S3
Results from LEfSe analysis showing taxa that significantly differ in abundance between the different analyzed species. Purple taxa significantly characterize the community of B. orientalis; pink taxon the community of B. japonicus; red taxon the community of C. pyrrhogaster; green taxon the community of O. splendida; and the blue taxa the community of R. japonica. (PDF 33.7 kb)
Supplementary Table S1
Core bacterial OTUs from the wild and captive individuals. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 11 kb)
Supplementary Table S2
Lower triangle reflects the pair-wise comparisons of the composition of the communities between species analyzed with PERMANOVA; P values are represented. Higher triangle reflects the distances between the communities: top value when determined with the Bray-Curtis distance matrix; bottom value when determined with the unweighted UniFrac distance matrix. (XLSX 9 kb)
Supplementary Table S3
Core bacterial OTUs from B. orientalis (Bom), B. japonicus (Buf), C. pyrrhogaster (Cyn), O. splendida (Odo), and R. japonica (Ran). Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 13 kb)
Supplementary Table S4
Core bacterial OTUs from the dorsal and ventral sides of B. orientalis. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 12 kb)
Supplementary Table S5
Core bacterial OTUs from the dorsal and ventral sides of B. japonicus. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 12 kb)
Supplementary Table S6
Core bacterial OTUs from the dorsal and ventral sides of C. pyrrhogaster. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 11 kb)
Supplementary Table S7
Core bacterial OTUs from the dorsal and ventral sides of E. andersoni. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 11 kb)
Supplementary Table S8
Core bacterial OTUs from the dorsal and ventral sides of O. splendida. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 12 kb)
Supplementary Table S9
Core bacterial OTUs from the dorsal and ventral sides of R. japonica. Abundance reflects the abundance of the OTU on the dataset and is in percentage. (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Sabino-Pinto, J., Bletz, M.C., Islam, M.M. et al. Composition of the Cutaneous Bacterial Community in Japanese Amphibians: Effects of Captivity, Host Species, and Body Region. Microb Ecol 72, 460–469 (2016). https://doi.org/10.1007/s00248-016-0797-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-016-0797-6