
Abstract
The patterns and drivers of bacterial strain dominance remain poorly understood in natural populations. Here, we cultured 1292 Bradyrhizobium isolates from symbiotic root nodules and the soil root interface of the host plant Acmispon strigosus across a >840-km transect in California. To investigate epidemiology and the potential role of accessory loci as epidemic drivers, isolates were genotyped at two chromosomal loci and were assayed for presence or absence of accessory “symbiosis island” loci that encode capacity to form nodules on hosts. We found that Bradyrhizobium populations were very diverse but dominated by few haplotypes—with a single “epidemic” haplotype constituting nearly 30 % of collected isolates and spreading nearly statewide. In many Bradyrhizobium lineages, we inferred presence and absence of the symbiosis island suggesting recurrent evolutionary gain and or loss of symbiotic capacity. We did not find statistical phylogenetic evidence that the symbiosis island acquisition promotes strain dominance and both symbiotic and non-symbiotic strains exhibited population dominance and spatial spread. Our dataset reveals that a strikingly few Bradyrhizobium genotypes can rapidly spread to dominate a landscape and suggests that these epidemics are not driven by the acquisition of accessory loci as occurs in key human pathogens.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A critical goal in bacteriology is to understand patterns of genotypic abundance and epidemic spread. Of particular interest are host-associated bacteria, including pathogens and symbionts. These diverse bacterial lineages colonize host surfaces, inhabit specific tissues or cells, and can often persist free in soils and or aquatic habitats between phases of host infection [1, 2]. The capacity of bacteria to thrive in host tissues is often modulated by the presence of plasmids and genomic islands, cassettes of accessory loci that can get transmitted among genomes. Horizontal transfer of these accessory loci often engenders bacteria with suites of fitness-enhancing traits including host infection capacity, pathogenicity, multidrug resistance, and metabolic flexibility [3–5]. The acquisition of plasmids and genomic islands has been implicated in epidemic outbreaks, including Pseudomonas aeruginosa, Staphylococcus aureus, and Yersinia spp. [6–8]. But in natural settings, we understand little about how bacterial strains vary in their capacity to dominate local sites or host populations or to spread among sites across ecological barriers. In particular, almost nothing is known about patterns of dominance and epidemic spread in symbiotic bacteria, which are important for human health, the success of crops, and other ecosystem services.
Rhizobia are proteobacteria that can exhibit capacity to infect leguminous plants and fix atmospheric nitrogen for their hosts [9]. Globally, rhizobia are responsible for the fixation of ~150 Tg of nitrogen per year [10] and their symbiosis with legumes represents the largest input source of nitrogen into terrestrial ecosystems [11]. In agriculture, legumes account for ~27 % of global crop production [12] and are valued for their capacity to grow in nitrogen depauperate soils. Similar to bacterial pathogens, rhizobia can acquire accessory DNA elements that confer the capability to colonize and infect hosts. Rhizobial genomes are subdivided into genome regions specific for their life stages, with chromosomal loci expressed during free-living phases in the soil and symbiosis loci expressed inside of host cells [13, 14]. Symbiosis loci required for host nodulation and nitrogen fixation are clustered onto large plasmids or genomic islands [15–19], that can be transferred among lineages, presumably via conjugation [20–22]. Non-nodulating rhizobia are also common [23, 24], and these strains often lack some or all of the characterized symbiosis loci [23–28].
Bradyrhizobium is the most cosmopolitan rhizobial lineage and is found free-living in soils and in aquatic environments as well as in symbiotic association with plant and animal hosts, including humans [29–37]. Recent work suggests that there are ~19 species of Bradyrhizobium that form symbiotic associations with legumes [36], although non-symbiotic strains are also common in soils [37]. Bradyrhizobium can nodulate diverse wild legumes as well as staple crops such as soybeans (Glycine), peanuts (Arachis), and cowpea (Vigna) [38, 39]. In the model genome, Bradyrhizobium diazoefficiens strain USDA110 (previously B. japonicum), symbiosis-specific genes are clustered within a 410-kb symbiosis island in which the G + C content differs from the rest of the genome [40]. Some divergent Bradyrhizobium spp. (e.g., BTAi1 and ORS278) lack a symbiosis island and use a different mechanism to nodulate hosts [41]. Previous studies have revealed that Bradyrhizobium populations can exhibit patterns consistent with epidemics such as a few genotypes existing at high frequency at a single site [33] or genotypes isolated at multiple locations [34, 35, 37]. But these patterns remain poorly understood, and it is unclear what role the symbiosis island might play as a driver of increased abundance and epidemic spread.
Here, we investigated the population genetic structure of Bradyrhizobium spp. cultured from Acmispon strigosus (formerly Lotus strigosus), a native annual legume common across the Pacific Southwest of the United States. We cultured 850 A. strigosus nodules from 14 natural sites across California encompassing 185 plants collected over a >840-km transect. In parallel, we isolated 442 root surface Bradyrhizobium from three focal host populations within this range, which includes isolates that lack symbiosis islands and cannot infect Acmispon hosts. All 1292 isolates were sequenced at two hypervariable chromosomal loci, and we used a combination of greenhouse inoculation assays and PCR to test for presence of the symbiosis island in all the root surface isolates. We assigned haplotypes and symbiotic capacity information to all isolates and examined the frequency and spatial spread of epidemic rhizobial genotypes within and among host populations. Our goals were to (i) investigate genotype frequency and spatial spread of Bradyrhizobium in native A. strigosus hosts, (ii) infer the presence or absence of the symbiosis island in Bradyrhizobium, (iii) test for the role of symbiosis island acquisition as a driver of Bradyrhizobium strain dominance, and (iv) test for community structure of rhizobial isolates due to other abiotic or biotic factors.
Methods
Collection of Bradyrhizobium Isolates
Bradyrhizobium was isolated from the nodules and the soil root interface of A. strigosus, and clonal cultures were grown and archived for genotyping following published protocols [33]. Briefly, whole plants were excavated from native sites and transported in sealed plastic bags to the laboratory where they were washed to remove soil with tap water and sterilized tools were used to remove root nodules. Nodules were surface sterilized with bleach and rinsed with sterile water before being crushed with glass rods and the contents plated on a modified arabinose gluconate medium (MAG) [33]. For root surface isolates, the roots were dissected into ~1-cm sections, were divided into root tips and “old” roots, and were vortexed in a sterile solution of 0.01 % Tween 20 (Fisher Scientific Fair Lawn, NJ). The wash solution was then serially diluted and plated on glucose-based rhizobium-defined medium (GRDM) with cyclohexamide as an antifungal and bromothymol blue as a pH indicator [33]. Among the resultant colonies, we selected for Bradyrhizobium based on growth rate, color, and ability to grow on MAG and GRDM but not on Luria–Bertani (LB) medium [33]. Plant hosts for culturing were collected from 14 field collection sites across California covering an ~840-km transect. Collection sites included University of California Natural Reserves (Bodega Marine Reserve, Burns Piñon Ridge Reserve, and Motte Rimrock Reserve); an undeveloped site in the hills above University of California–Riverside, a biological field station in Claremont, CA (Robert J. Bernard Biological Field Station); natural preserves (Madrona Marsh Preserve, Pismo Dunes Natural Preserve, and Whitewater Preserve); a wildlife refuge (Guadalupe–Nipomo Dunes National Wildlife Refuge); two separate sites within a large state park (Anza Borrego Desert State Park); a municipal park (Griffith Park); a site adjacent to the San Dimas Reservoir; and an undeveloped site adjacent to human development (San Dimas Canyon) (Table S1). Nodule isolates were collected from plants at all sites, but root surface isolates were only collected from plants at the Bodega Marine Reserve, Motte Rimrock Reserve, and the undeveloped site in the hills above the University of California–Riverside.
Sequencing and Haplotype Analysis
Partial sequences from two chromosomal loci, glutamine synthetase (glnII) and recombinase (recA), totaling ~1 kb were PCR amplified and sequenced at the Institute for Integrative Genome Biology of UC Riverside using published protocols [34]. Only sequences with unambiguous bases were utilized leading to a total of 1292 sequenced isolates. Sequences from each locus were aligned separately using Clustal Omega, and isolates with identical sequences for each locus were determined using the “find redundant” command within the MacClade program [42]. Unique sequences, or haplotypes, were defined for each locus separately and for the concatenated dataset. Abundance was calculated for the concatenated dataset as the number of times each haplotype was isolated.
Symbiotic Capacity Assessment
We conducted a combination of assays on root surface isolates to test for symbiotic capacity. A subset of isolates (75) were previously assessed using greenhouse nodulation assays on A. strigosus, which has already been shown to be a permissive host on diverse Bradyrhizobium [24, 43, 44]. Here, we conducted greenhouse nodulation experiments on an additional 55 isolates, using identical procedures. Briefly, at least five seedlings per tested Bradyrhizobium isolate were grown in sterile conditions and were inoculated clonally with 5 × 108 cells, and parallel control hosts were inoculated with sterile water. At 8-week postinoculation, all hosts were unpotted, roots and shoots were weighed, and roots were checked for nodules. In all cases, controls lacked nodules. Hosts given the same inoculated strains either all became nodulated or were all lacking nodules. The remaining 342 root surface isolates were classified as symbiotic or non-symbiotic based on success or failure of PCR amplification of at least one symbiosis island locus (nifD, nodD-A, nodZ, and nolL) [22, 24, 45]. Earlier analyses showed that successful amplification of these loci, giving a band of the correct size, is a reliable indicator of presence of the symbiosis island [24]. Many isolates were tested at two or more loci (160/342; Table S1).
Phylogenetic Reconstruction and Species Designation
Phylogenies were reconstructed using the concatenated glnII and recA sequences from the cultured isolates and from the following reference strains: Bradyrhizobium arachidis (CCBAU33067), B. betae (PL7HG1), B. canariense (SEMIA928), B. cytisi (LMG25866), B. diazoefficiens (SEMIA5080), B. elkanii (USDA46), B. iriomotense (EK05), B. diazoefficiens (USDA110), B. lablabi (CCBAU61434), B. liaoningense (SEMIA5025), B. retamae (Ro19), and B. yuanmingense (R2m). Reference strains were chosen to represent all known species of Bradyrhizobium that aligned fully with our sequenced glnII and recA regions (National Center for Biotechnology Information (NCBI) as of November 18, 2014), and Mesorhizobium loti (MAFF303099) was used as an outgroup. All sequences were blasted against the reference strains to confirm their identity as Bradyrhizobium spp. The GTR model of evolution was selected from the Akaike information criterion in jModelTest2 [46], and the phylogenetic tree was reconstructed in PhyML 3.0 [47] utilizing a BioNJ starting tree and subtree pruning and regrafting (SPR). Branch support was estimated using the fast approximate likelihood ratio test (aLRT) and the Shimodaira–Hasegawa-like (SH-like) procedure [48]. To be consistent with other studies, Bradyrhizobium species were defined as the monophyletic clades including no more than one type strain with branch support ≥0.90 [49] and attempting to adhere to past species demarcations that utilized some of the same loci [35]. We analyzed inter-species variation using the ratio of fixed to shared polymorphisms using DnaSP [50].
Sequence Statistics
Using the concatenated dataset, we calculated strain richness (number of unique haplotypes/number of isolates) and strain dominance (abundance of each haplotype/number of isolates) analogues of species richness and evenness [51]. For each of the 14 field collection sites, haplotypes were defined as dominant if they were collected at least five times and represented at least 10 % of the total isolates at that site. Spatial spread was defined as the maximum distance between any individual isolates with the same haplotype. GPS coordinates for distances used the midpoint of each of the 14 field collection sites, because distances within sites were small compared to between site differences. We also calculated Hd (haplotype diversity—probability that two haplotypes drawn uniformly at random from the population are not the same), π (nucleotide diversity—average number of nucleotide differences per site between two sequences), k (average number of nucleotide differences), linkage disequilibrium (average absolute D′), recombination (R), and the minimum number of recombination events [52–56] using DnaSP [50].
Trait Analysis
We tested for significant phylogenetic signal for the traits of symbiotic capacity, abundance, and spatial spread, which is a prerequisite for quantitative analysis of traits in a phylogenetic framework. We used Pagel’s lambda, estimated with the “fit discrete” function in the “Geiger” package [57], and for symbiotic capacity, we also used Fritz Purvis’ D, which was estimated using the “phylo.d” function in the “Caper” package [58]. The Mk1 model of maximum likelihood as well as parsimony were used for ancestral state reconstruction of symbiotic capacity with a modified phylogenetic tree in Mesquite [59]. Duplicate taxa were added to the phylogenetic tree whenever a single haplotype encompassed both symbiotic and non-symbiotic isolates to avoid the ambiguity of multiple character states being assigned to a single taxon (i.e., haplotype). We tested for correlated evolution between symbiotic capacity and haplotype abundance with the phy.anova command in the Geiger package in R [57]. We took several steps to avoid bias caused by sampling in this analysis. Firstly, we used the subset of isolates collected from plants where both root surface and nodule collections had been made (to avoid sampling bias). Secondly, among these plants, we equally sampled across the whole root system representing samples from nodules and from the soil-root interface with equal probability per sampled root. The resultant dataset included 442 root surface isolates and 116 nodule isolates from three field locales. We also used a standard ANOVA in JMP [60] to examine variation between symbiotic and non-symbiotic isolates in terms of abundance and spatial spread. This latter analysis does not take phylogenetic relationships into account and thus assumes that trait data are independent of strain relatedness.
Community Structure
We analyzed isolation by distance with a Mantel test correlating F st and physical distance matrices within PASSaGE [61]. We used Fast UniFrac [62] to test for significant differentiation among Bradyrhizobium communities at different collection sites. The “cluster sample” tool was used to cluster the collection sites based on the phylogenetic lineages they contained, and the “jackknife sample cluster” tool was used to assess confidence in the collection site clusters. We employed the “sample distance matrix” to numerically compare distances among collection sites. Abundance was incorporated into Fast UniFrac analyses whenever possible. The jackknife analysis was performed with the number of sequences kept equal to the smallest sample size with 100 permutations. We used the “exact test of population differentiation” in Arlequin [63] to assess differentiation among collection sites and to investigate other drivers of Bradyrhizobium community structure including root isolate types (nodule, root-tip surface, and old root surface), symbiotic statuses (symbiotic and non-symbiotic), and collection year.
Results
Haplotype Designation, Abundance, and Spatial Spread
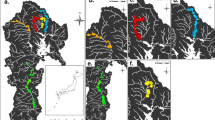
The 1292 concatenated glnII and recA sequences resulted in 290 haplotypes, all of which were classified as Bradyrhizobium. Most of the haplotypes were unique (isolated a single time, 184/290; Table S1). Among the remaining haplotypes, 13 were defined as dominant in at least one site and these 13 haplotypes constituted the majority of collected isolates (706/1292). We found dominant haplotypes at all but the least sampled field collection site (Anza Borrego Desert State Park–Palm Canyon; Table 1). Most haplotypes (257/290) were only found at a single collection site. Conversely, among the dominant haplotypes, most were also found to be epidemic (7/13; collected at sites spanning ≥10 km). Spatial spread for epidemic haplotypes varied from ~100 to 750 km, and we collected epidemic haplotypes at all but the two least sampled sites (Anza Borrego Desert State Park–Palm Canyon and Pismo Dunes Natural Preserve) (Table 1). One epidemic haplotype (G03_R01) encompassed 27 % of all isolates collected (355 isolates) and was found at all but four collection sites (Pismo Dunes Natural Preserve, Griffith Park, and Anza Borrego State Park–Road/Palm Canyon sites; Fig. 1).

Map of California indicates collection sites with black dots. Pie charts connected to black dots illustrate the relative frequencies of five focal haplotypes. The five haplotypes chosen incorporate the four haplotypes with the highest abundance and the four haplotypes with the greatest spatial spread. The distribution of epidemic haplotype G03_R01 is hypothesized based on presence or absence of data from all collection sites. Starting from the upper left and moving clockwise, the collection sites are Bodega Marine Reserve, San Dimas Canyon, San Dimas Reservoir, Bernard Field Station, UC Riverside, Motte Rimrock Reserve, Whitewater, Burns Pinon Ridge, Anza Borrego Canyon, Anza Borrego Roadsize, Madrona Marsh, Griffith Park, Nipomo Dunes, and Pismo Dunes
Symbiotic Capacity Assessment
Using the combined dataset of greenhouse inoculation assays and PCR assays, we inferred 886 isolates to be symbiotic and 406 to be non-symbiotic (Table S1). For ~2 % of the 1292 isolates, there was at least one piece of conflicting information about symbiotic capacity, most often including nodule isolates that failed to amplify one or more of the four symbiosis island loci (17) but also including conflicting results between PCR amplification assays (9) (Table S1).
Most dominant haplotypes only encompassed isolates that were inferred to be symbiotic, but a single dominant haplotype (G64_R29) only had non-symbiotic isolates. Of the dominant haplotypes that included both symbiotic and non-symbiotic isolates, mean abundance was higher for symbiotic (36) versus non-symbiotic isolates (15.6) but the difference was not significant (t = 1.48, df = 4, p = 0.214). Among the epidemic haplotypes, most encompassed symbiotic and non-symbiotic isolates (5/7), with symbiotic isolates being more frequent on average than non-symbiotic ones, but without a significant difference (90.6 vs 18.4; t = 2.05, df = 4, p = 0.110).
Phylogenetic Analysis
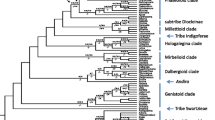
The Bradyrhizobium phylogeny encompassed 20 deeply diverged, monophyletic lineages (clades) that are consistent with species-level divergence, including six that were previously identified (B. betae, B. canariense, B. cytisi, B. liaoningense, and B. retamae) and 14 clades that did not encompass one of the type strains (Figs. 2 and S1). Most (161) comparisons between these 20 clades uncovered more fixed than shared polymorphisms, also consistent with species designation. Two comparisons had the same number of fixed and shared polymorphisms, and 27 had more shared than fixed polymorphisms (Table S2). Almost half of these clades (8/20) were only collected at a single site. However, nearly all collection sites (13/14) were inhabited by multiple clades of Bradyrhizobium (Table S3). Bradyrhizobium canariense was particularly widespread and was collected at 11 sites. Population genetic statistics were analyzed for all Bradyrhizobium clades that encompassed multiple isolates (Table 2). Linkage was high between all SNPs for all clades (>0.9). Strain richness, Hd, π, and recombination varied widely between clades, likely because of variation in sampling. When only clades with over 40 isolates were assessed, Hd, π, and recombination were comparable.

PhyML 3.0 phylogenetic tree reconstructed from concatenated glutamine synthetase (glnII) and recombinase (recA) loci. Non-symbiotic taxa are indicated with red dots and symbiotic taxa with black dots. Inferred ancestral states of non-symbiotic and symbiotic are indicated by red and black branches, respectively, and ambiguous nodes are colored grey (estimated likelihood proportion was <66.66 %). The relative abundance of a haplotype and the spatial spread are indicated by stacked blue and green bars. Major clades are indicated with brackets. Reference strains can be identified by the lack of symbiotic capacity, abundance, and spatial spread data. The strains include (1) Mesorhizobium loti MAFF303099, (2) Bradyrhizobium retamae Ro19, (3) Bradyrhizobium elkanii USDA46, (4) Bradyrhizobium lablabi CCBAU61434, (5) B. diazoefficiens SEMIA5080, (6) B. diazoefficiens (japonicum) USDA110, (7) Bradyrhizobium betae PL7HG1, (8) Bradyrhizobium iriomotense EK05, (9) Bradyrhizobium arachidis CCBAU33067, (10) Bradyrhizobium yuanmingense R2m, (11) Bradyrhizobium liaoningense SEMIA5025, (12) Bradyrhizobium cytisi LMG25866, and (13) Bradyrhizobium canariense SEMIA928
Ancestral state reconstructions were similar for parsimony and likelihood models (Figs. 2 and S2), and both infer gains and or losses of symbiotic capacity across multiple Bradyrhizobium clades (Table S4). Because many of the nodes on the phylogenetic tree are poorly resolved, it is impossible to estimate the number of gain or loss events with confidence. Considering only the 20 deeply diverged clades with likelihood support values >0.90, 10 of them encompassed both symbiotic and non-symbiotic isolates consistent with multiple gain and loss events (Fig. S1). Using both parsimony and a conservative maximum likelihood estimate (likelihood decision threshold of 2.0), we inferred about 1.5–2× as many loss events as gains (Table S4).
Symbiotic capacity exhibited significant phylogenetic signal, but abundance and spatial spread did not (Table S5). When we assessed symbiotic capacity and abundance for correlated evolution using chi-squared tests in JMP, we found a significant positive correlation; hence, that symbiotic clades on average exhibit higher abundance than non-symbiotic clades. When we tested for correlated evolution of these traits, we did not find a relationship (Tables S6 and S7); hence, that acquisition of a symbiosis island is not statistically associated with increase in haplotype abundance. Moreover, we did not find evidence for correlated evolution of symbiotic capacity with spatial spread using either the chi-squared tests in JMP or the phy.anova in Geiger.
Community Analyses
We found significantly different Bradyrhizobium communities among most collection sites (Table S8), among different isolate types (nodule, root-tip surface, and old root surface), symbiotic statuses (symbiotic and non-symbiotic), and collection years (Table S9). Non-symbiotic populations were significantly differentiated from isolates that were symbiotic within the same site at the Bodega Marine Reserve and Motte Rimrock reserve (Table S10). Two of four sites that were sampled multiple times exhibited population differentiation from year to year, San Dimas Canyon and Burns Piñon Ridge Reserve (Table S11). We did not find evidence for isolation by distance using the Mantel test. This is supported by the Fast UniFrac analyses in which clustering closely follows species makeup rather than geographical location (Figs. S3 and S4).
Discussion
Our study uncovered an epidemic distribution of Bradyrhizobium haplotypes across California with a striking divide between rare and dominant haplotypes. Although we recovered 290 haplotypes, the majority of isolates were encompassed within 13 dominant haplotypes (707/1292). Among the dominant haplotypes, six were endemic and seven were epidemic including haplotype G03_R01 that was dominant at most sites (10/14), exhibited a spatial spread of 750 km, and constituted nearly 30 % of the total isolates (355/1292) (Table 1). Several studies have uncovered rhizobial genotypes that achieve high frequency at localized sites [33, 51, 64], but haplotype G03_R01 dominates both locally and also across major ecological boundaries including mountain ranges and areas of inhospitable habitat. It is striking that haplotype G03_R01 was found in sites that vary markedly in their patterns of rainfall, temperature, plant community, and key soil nutrients [65, 66], especially given that most of the Bradyrhizobium lineages that we uncovered were relatively localized in their distribution. Recent work that focused on non-symbiotic lineages of Bradyrhizobium in soils has also found evidence of widespread genotypes, suggesting that this might be a common pattern for these bacteria [37].
We uncovered a broad diversity of Bradyrhizobium species nodulating and inhabiting the root surfaces of A. strigosus. The most recent efforts in defining symbiotic lineages of Bradyrhizobium propose 19 species [36]. Using a conservative species definition (monophyletic clades including no more than one type strain with branch support ≥0.90), we recovered 20 species including 14 lineages that were not represented in the NCBI database using these common loci (Fig. S1). No single method has been agreed upon for defining species of bacteria, but most definitions use phylogenetic clusters of multiple conserved loci, as we did here [67]. Despite the diversity that we uncovered, six out of the seven epidemic haplotypes are found within the B. canariense and B. novel I species (Table 2). Important differences can exist between rhizobial lineages including the diversity of host plants infected, metabolite utilization, and antibiotic resistance [68–70], and these sources of variation may be driving differences in epidemic distributions among Bradyrhizobium taxa. We also found that Bradyrhizobium communities from different collection sites varied in species diversity (Table S3). We found the greatest diversity at the three sites where we collected root surface samples, consistent with previous work uncovering greater genotypic diversity among root surface isolates as opposed to nodule populations [33].
Genomic island acquisition can facilitate novel bacterial traits that confer fitness benefits in an environment specific manner [71]. However, we did not find support for the hypothesis that symbiosis island acquisition is an evolutionary driver of strain abundance or spatial spread in Bradyrhizobium (Table S7). Instead, we uncovered epidemics of only a few extremely abundant symbiotic haplotypes within B. canariense, and in this clade, we found evidence consistent with multiple events of symbiosis island gain and loss. Our dataset shows that both acquisition and loss of the symbiosis island appear to occur frequently across the sampled Bradyrhizobium populations. Both maximum likelihood and parsimony analyses suggest many more loss than gain transitions. Many types of mutations can result in the conversion of symbiotic strain to a non-symbiotic. But only the whole-scale horizontal transmission of the symbiosis island has been associated with gain of nodulation and nitrogen fixation in Bradyrhizobium. Given the more frequent loss events, some additional diversification must occur in the symbiotic lineages relative to the lineages without symbiosis islands.
The capacity to infect A. strigosus appears to have a significant affect on structuring Bradyrhizobium populations. We found support for differentiation in 2/3 comparisons of symbiotic and non-symbiotic communities (Table S10), and we also found that 8/9 comparisons of Bradyrhizobium communities collected from different plant parts exhibited differentiation (Table S9). We also found evidence for local differences among collection sites that could be responsible for Bradyrhizobium population differentiation; however, drift cannot be ruled out by these analyses (Table S8). Finally, we found evidence for temporal differentiation in 3/5 comparisons (Table S11). Taken together, these data are consistent with previous evidence showing that plant and soil factors affect population structure in the rhizosphere [72].
In summary, our analysis found that native Bradyrhizobium populations across California are dominated by a very small handful of haplotypes. We found that on average, symbiotic strains are more common than non-symbiotic ones and that symbiotic strains are also more likely to be dominant and to spread spatially. Nonetheless, we found evidence of both symbiotic and non-symbiotic strains achieving high abundance and spreading across great distances. Our ancestral state reconstruction inferred that the symbiosis island has been repeatedly gained and lost across the A. strigosus sampling range, and we did not find support for the hypothesis that acquisition of symbiosis islands serves as a driver of strain dominance or spread. We suggest that traits encoded on the Bradyrhizobium chromosome, that expresses traits important for soil survival [13, 14], are most likely responsible for variation in the capacity to spread epidemically.
References
Bright M, Bulgheresi S (2010) A complex journey: transmission of microbial symbionts. Nat Rev Microbiol 8:218–230. doi:10.1038/nrmicro2262
Sachs JL, Skophammer RG, Bansal N, Stajich JE (2013) Evolutionary origins and diversification of proteobacterial mutualists. Proc R Soc B Biol Sci 281:20132146–20132146. doi:10.1098/rspb.2013.2146
Groisman EA, Ochman H (1996) Pathogenicity islands: bacterial evolution in quantum leaps. Cell 87:791–794. doi:10.1016/S0092-8674(00)81985-6
Jain R, Rivera MC, Moore JE, Lake JA (2003) Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol 20:1598–1602. doi:10.1093/molbev/msg154
Gal-Mor O, Finlay BB (2006) Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol 8:1707–1719. doi:10.1111/j.1462-5822.2006.00794.x
Bach S (2000) The Yersinia high-pathogenicity island is present in different members of the family Enterobacteriaceae. FEMS Microbiol Lett 183:289–294. doi:10.1016/S0378-1097(00)00005-7
Diep BA, Gill SR, Chang RF et al (2006) Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367:731–739. doi:10.1016/S0140-6736(06)68231-7
Qiu X, Gurkar AU, Lory S (2006) Interstrain transfer of the large pathogenicity island (PAPI-1) of Pseudomonas aeruginosa. Proc Natl Acad Sci 103:19830–19835. doi:10.1073/pnas.0606810104
Sprent JI (2001) Nodulation in legumes. Royal Botanic Gardens, Kew
Sugawara M, Epstein B, Badgley BD et al (2013) Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol 14:R17. doi:10.1186/gb-2013-14-2-r17
Cleveland CC, Townsend AR, Schimel DS et al (1999) Global patterns of terrestrial biological nitrogen (N2) fixation in natural ecosystems. Glob Biogeochem Cycles 13:623–645. doi:10.1029/1999GB900014
Graham PH, Vance CP (2003) Legumes: importance and constraints to greater use. Plant Physiol 131:872–877. doi:10.1104/pp. 017004
Uchiumi T, Ohwada T, Itakura M et al (2004) Expression islands clustered on the symbiosis island of the Mesorhizobium loti genome. J Bacteriol 186:2439–2448. doi:10.1128/JB.186.8.2439-2448.2004
Pessi G, Ahrens CH, Rehrauer H et al (2007) Genome-wide transcript analysis of Bradyrhizobium japonicum bacteroids in soybean root nodules. Mol Plant Microbe Interact 20:1353–1363. doi:10.1094/MPMI-20-11-1353
Kaneko T (2000) Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res 7:331–338. doi:10.1093/dnares/7.6.331
Galibert F (2001) The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293:668–672. doi:10.1126/science.1060966
Kaneko T, Nakamura Y, Sato S et al (2002) Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res 9:189–197. doi:10.1093/dnares/9.6.189
Young JPW, Crossman LC, Johnston AWB et al (2006) The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7:R34. doi:10.1186/gb-2006-7-4-r34
Lee K-B, De Backer P, Aono T et al (2008) The genome of the versatile nitrogen fixer Azorhizobium caulinodans ORS571. BMC Genomics 9:271. doi:10.1186/1471-2164-9-271
Young JPW (1996) Diversity and phylogeny of rhizobia. New Phytol 133:87–94
Perret X, Staehelin C, Broughton WJ (2000) Molecular basis of symbiotic promiscuity. Microbiol Mol Biol Rev 64:180–201. doi:10.1128/MMBR.64.1.180-201.2000
Moulin L, Béna G, Boivin-Masson C, Stępkowski T (2004) Phylogenetic analyses of symbiotic nodulation genes support vertical and lateral gene co-transfer within the Bradyrhizobium genus. Mol Phylogenet Evol 30:720–732. doi:10.1016/S1055-7903(03)00255-0
Segovia L, Piñero D, Palacios R, Martínez-Romero E (1991) Genetic structure of a soil population of Rhizobium leguminosarum. Appl Environ Microbiol 57:426–433
Sachs JL, Ehinger MO, Simms EL (2010) Origins of cheating and loss of symbiosis in wild Bradyrhizobium. J Evol Biol 23:1075–1089. doi:10.1111/j.1420-9101.2010.01980.x
Sullivan JT, Eardly BD, van Berkum P, Ronson CW (1996) Four unnamed species of nonsymbiotic rhizobia isolated from the rhizosphere of Lotus corniculatus. Appl Environ Microbiol 62:2818–2825
Saito A, Mitsui H, Hattori R et al (1998) Slow-growing and oligotrophic soil bacteria phylogenetically close to Bradyrhizobium japonicumin. FEMS Microbiol Ecol 25:277–286. doi:10.1111/j.1574-6941.1998.tb00480.x
Pongsilp N, Teaumroong N, Nuntagij A et al (2002) Genetic structure of indigenous non-nodulating and nodulating populations of Bradyrhizobium in soils from Thailand. Symbiosis 33:39–58
Okubo T, Tsukui T, Maita H et al (2012) Complete genome sequence of Bradyrhizobium sp. S23321: insights into symbiosis evolution in soil oligotrophs. Microbes Environ JSME 27:306–315
Bhatt AS, Freeman SS, Herrera AF et al (2013) Sequence-based discovery of Bradyrhizobium enterica in cord colitis syndrome. N Engl J Med 369:517–528. doi:10.1056/NEJMoa1211115
Chaintreuil C, Giraud E, Prin Y et al (2000) Photosynthetic bradyrhizobia are natural endophytes of the African wild rice Oryza breviligulata. Appl Environ Microbiol 66:5437–5447. doi:10.1128/AEM.66.12.5437-5447.2000
Costello EK, Carlisle EM, Bik EM, et al. (2013) Microbiome assembly across multiple body sites in low-birthweight infants. mBio 4:e00782–13–e00782–13. doi: 10.1128/mBio.00782-13
Hunt KM, Foster JA, Forney LJ et al (2011) Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS ONE 6, e21313. doi:10.1371/journal.pone.0021313
Sachs JL, Kembel SW, Lau AH, Simms EL (2009) In situ phylogenetic structure and diversity of wild Bradyrhizobium communities. Appl Environ Microbiol 75:4727–4735. doi:10.1128/AEM.00667-09
Vinuesa P, Silva C, Werner D, Martínez-Romero E (2005) Population genetics and phylogenetic inference in bacterial molecular systematics: the roles of migration and recombination in Bradyrhizobium species cohesion and delineation. Mol Phylogenet Evol 34:29–54. doi:10.1016/j.ympev.2004.08.020
Vinuesa P, Rojas-Jimenez K, Contreras-Moreira B et al (2008) Multilocus sequence analysis for assessment of the biogeography and evolutionary genetics of four Bradyrhizobium species that nodulate soybeans on the Asiatic continent. Appl Environ Microbiol 74:6987–6996. doi:10.1128/AEM.00875-08
Azevedo A, Martins-Lopes F, Silla RP, Hungria M (2015) A database for the taxonomic and phylogenetic identification of the genus Bradyrhizobium using multilocus sequence analysis. BMC Genomics 16:S10
VanInsberghe D, Maas KR, Cardenas E et al (2015) Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. doi:10.1038/ismej.2015.54
Rivas R, Martens M, de Lajudie P, Willems A (2009) Multilocus sequence analysis of the genus Bradyrhizobium. Syst Appl Microbiol 32:101–110. doi:10.1016/j.syapm.2008.12.005
Parker MA (2015) The spread of Bradyrhizobium lineages across host legume clades: from Abarema to Zygia. Microb Ecol. doi:10.1007/s00248-014-0503-5
Göttfert M, Röthlisberger S, Kündig C et al (2001) Potential symbiosis-specific genes uncovered by sequencing a 410-kilobase DNA region of the Bradyrhizobium japonicum chromosome. J Bacteriol 183:1405–1412. doi:10.1128/JB.183.4.1405-1412.2001
Giraud E, Moulin L, Vallenet D et al (2007) Legumes symbioses: absence of Nod genes in photosynthetic bradyrhizobia. Science 316:1307–1312. doi:10.1126/science.1139548
Maddison WP, Maddison DR (2005) MacClade: analysis of phylogeny and character evolution
Sachs JL, Russell JE, Hollowell AC (2011) Evolutionary instability of symbiotic function in Bradyrhizobium japonicum. PLoS ONE 6, e26370. doi:10.1371/journal.pone.0026370
Ehinger M, Mohr TJ, Starcevich JB et al (2014) Specialization-generalization trade-off in a Bradyrhizobium symbiosis with wild legume hosts. BMC Ecol 14:8. doi:10.1186/1472-6785-14-8
Parker M (2000) Divergent Bradyrhizobium symbionts on Tachigali versicolor from Barro Colorado Island, Panama. Syst Appl Microbiol 23:585–90
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi:10.1038/nmeth.2109
Guindon S, Dufayard JF, Lefort V et al (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi:10.1093/sysbio/syq010
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55:539–552. doi:10.1080/10635150600755453
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. doi:10.1093/bioinformatics/btp187
Mcinnes A (2004) Structure and diversity among rhizobial strains, populations and communities—a review. Soil Biol Biochem 36:1295–1308. doi:10.1016/j.soilbio.2004.04.011
Lewontin RC (1964) The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49:49–67
Tajima F (1983) Evolutionary relationship of DNA sequences in finite populations. Genetics 105:437–460
Hudson RR, Kaplan NL (1985) Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164
Hudson RR (1987) Estimating the recombination parameter of a finite population model without selection. Genet Res 50:245–250
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Harmon LJ, Weir JT, Brock CD et al (2007) GEIGER: investigating evolutionary radiations. Bioinformatics 24:129–131. doi:10.1093/bioinformatics/btm538
Orme D (2012) The caper package: comparative analysis of phylogenetics and evolution in R
Maddison, W. P., Maddison, D. R. (2011) Mesquite
SAS Institute Inc (1989) JMP. SAS Institute Inc., Cary, NC
Rosenberg MS, Anderson CD (2011) PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2: PASSaGE. Methods Ecol Evol 2:229–232. doi:10.1111/j.2041-210X.2010.00081.x
Hamady M, Lozupone C, Knight R (2010) Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4:17–27. doi:10.1038/ismej.2009.97
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. doi:10.1111/j.1755-0998.2010.02847.x
Silva C, Eguiarte LE, Souza V (1999) Reticulated and epidemic population genetic structure of Rhizobium etli biovar phaseoli in a traditionally managed locality in Mexico. Mol Ecol 8:277–287. doi:10.1046/j.1365-294X.1999.00564.x
Baldwin BG, Goldman DH (2012) The Jepson manual: vascular plants of California, 2nd edn. University of California Press, Berkeley, Calif
Regus JU, Gano KA, Hollowell AC, Sachs JL (2014) Efficiency of partner choice and sanctions in Lotus is not altered by nitrogen fertilization. Proc R Soc B Biol Sci 281:20132587–20132587. doi:10.1098/rspb.2013.2587
Thompson CC, Amaral GR, Campeao M et al (2015) Microbial taxonomy in the post-genomic era: rebuilding from scratch? Arch Microbiol 197:359–370. doi:10.1007/s00203-014-1071-2
Dupuy N, Willems A, Pot B et al (1994) Phenotypic and genotypic characterization of bradyrhizobia nodulating the leguminous tree Acacia albida. Int J Syst Bacteriol 44:461–473. doi:10.1099/00207713-44-3-461
Geurts R (1996) Signal transduction in Rhizobium-induced nodule formation. Plant Physiol 112:447–453. doi:10.1104/pp. 112.2.447
Hollowell AC, Gano KA, Lopez G et al (2015) Native California soils are selective reservoirs for multidrug-resistant bacteria. Environ Microbiol Rep. doi:10.1111/1758-2229.12269
Dobrindt U, Hochhut B, Hentschel U, Hacker J (2004) Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2:414–424. doi:10.1038/nrmicro884
Berg G, Smalla K (2009) Plant species and soil type cooperatively shape the structure and function of microbial communities in the rhizosphere: plant species, soil type and rhizosphere communities. FEMS Microbiol Ecol 68:1–13. doi:10.1111/j.1574-6941.2009.00654.x
Acknowledgments
The following grants supported this study: to ACH a Herbert Kraft Scholarship and a UC Riverside Graduate Research Mentorship Fellowship and to JLS NSF DEB 0816663 and NSF DEB 1150278.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hollowell, A.C., Regus, J.U., Gano, K.A. et al. Epidemic Spread of Symbiotic and Non-Symbiotic Bradyrhizobium Genotypes Across California. Microb Ecol 71, 700–710 (2016). https://doi.org/10.1007/s00248-015-0685-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0685-5