
Abstract
Anaerobic digestion is an alternative method for the treatment of animal manure and wastewater. The anaerobic bioconversion of biomass requires a multi-step biological process, including microorganisms with distinct roles. The diversity and composition of microbial structure in pilot-scale anaerobic digestion operating at ambient temperature in Brazil were studied. Influence of the seasonal and temporal patterns on bacterial and archaeal communities were assessed by studying the variations in density, dynamic and diversity and structure. The average daily biogas produced in the summer and winter months was 18.7 and 16 L day−1, respectively, and there was no difference in the average methane yield. Quantitative PCR analysis revealed that no differences in abundances and dynamics were found for bacterial communities and the total number of Archaea in different seasons. Analysis of bacterial clone libraries revealed a predominance of Firmicutes (54.5 %/summer and 46.7 %/winter) and Bacteroidetes (31.4 %/summer and 44.4 %/winter). Within the Archaea, the phylum Euryarchaeota was predominant in both digesters. Phylogenetic distribution showed changes in percentage between the phyla identified, but no alterations were recorded in the quality and amount of produced methane or community dynamics. The results may suggest that redundancy of microbial groups may have occurred, pointing to a more complex microbial community in the ecosystem related to this ambient temperature system.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The scale of dairy farming operations has increased significantly due to the high demand of dairy products for consumption. Consequently, large quantities of cattle manure are produced, leading to the contamination of multiple environments [1]. Anaerobic digestion is an alternative method for the treatment of animal manure and wastewater, as the process combines waste treatment and renewable energy recovery [2]. Currently, anaerobic digestion has been widely applied for the production of a clean fuel from feed stocks, such as animal manure, in many countries [3].
During the anaerobic bioconversion of biomass to carbon dioxide and methane, a multi-step biological process is required, including hydrolytic, fermentative, acetogenic and methanogenic microorganisms, which play distinct roles in digesting many different types of organic wastes [4]. The abundance and composition of the microbial community in a treatment process is complex and directly determines its efficiency and biogas yield [5]. According to the literature, microbial ecosystems may be directly modulated by biotic and abiotic characteristics, such as, in biodigesters, by substrate. In addition, the operational management may also influence microbial structure [2].
Brazil is a country that presents climatic conditions in the mesophilic range (between 20 and 40 °C) that ensures anaerobic digestion at ambient temperature throughout the year, without digester heating [6]. In this regard, the positive use of this treatment of cattle manure and wastewater at ambient temperatures would be a significant success that could have a large effect on reducing the cost and energy requirements. However, there is temperature variation between the seasons (about 10 °C) and it is not known whether this variation in the mesophilic range can affect the stability of the microbial community and consequently the production of biogas [7, 8]. Few studies have been carried out to investigate the effect of temperature shift on the microbial community structure in biogas reactors operating under ambient temperature, especially those in anaerobic digesters that treat dairy manure [9–11].
Knowledge of the dynamics and composition of the microbial community in this context is fundamental for understanding their function in waste degradation and achievement of the stable operation of the process. In this study, the temporal and seasonal variability of bacterial and archaeal communities in two ambient temperature pilot-scale biodigesters were investigated by 16S ribosomal RNA (rRNA) gene-targeted PCR amplification and single-strand conformation polymorphism (CE-SSCP) analysis, followed by 16S rRNA sequencing analysis.
Material and Methods
Pilot-Scale Biogas Reactor
The microbial community structure was analysed in two pilot-scale tubular digesters operating ambient temperatures for different seasons, with a 60-day retention time, and 60-L working volume. The pilot continuous digesters consist of PVC containers, without heating or agitation. Each digester is connected to a gas metre, as shown in Fig. 1. The gas metre consists of two PVC vessels, the one (25-cm diameter) placed upside down inside the other (30-cm diameter). The biogas line from the digester was connected to this tube. The cylinder was then filled with water, up to the height of the PVC tube. Biogas production is measured by the displacement of the plastic vessel. By knowing this height (displacement) and the diameter of the plastic cylinder, the volume of biogas produced is calculated. Temperature and pH was monitored in the liquid phase.

Details of a tubular continuous anaerobic digester, gasometer and different equipment used to perform the tests
Fresh dairy cattle manure was collected weekly from the experimental Embrapa dairy cattle field located in Coronel Pacheco city, Minas Gerais state, Brazil, and transported to the laboratory within 1 h of sampling. On arrival at the laboratory, the manure was chilled to 4 °C to reduce die-off of bacteria and to allow it to be used for up to 6 days. Before being used as a substrate in the pilot-scale biodigesters, mixtures were prepared daily from the stored wastes, maintaining the weight ratio of each residue in the mixture and completing the final volume with cattle wastewater to obtain a total solid content of 4–5 % and added to each digester an amount of 2 L.
Parameters such as total solids (TS), volatile solids (VS), total chemical oxygen demand (TCOD), soluble chemical oxygen demand (SCOD), and pH of influent and effluent samples were estimated as per standard methods [12].
The samples were collected equally during two different periods: between January to March (2012) (summer) and June to July (2012) (winter). The pH value of the cattle manure (influent) was between 6.55 and 6.86, and the loading rates were around 13.3 g mL−1 of VS in the summer and 12.4 g mL−1 of VS in the winter months. Biogas composition was analysed every week by gas chromatography (Agilent Technologies, 7820A) equipped with two columns: HP-Molsieve 19095P-MS6 and HP-PLOT/Q 19095P-QO4, employing thermal conductivity detection where helium was used as carrier gas. The standard calibration curve was made with gas mixtures containing CH4 at three levels covering the range of 20–99.999 % and CO2 at four levels (range of 20–79.9 %). All measurements were made in triplicate and the averages were taken as the representative values.
DNA Extraction
Samples were identified according to season, day of collection and source (two digesters): samples collected during the summer and winter were denoted with the letters S (Summer) and W (Winter), respectively; the numbers (0 to 60) were added according to the sampling days during anaerobic digestion, the letters (a and b) correspond to each of the digesters where the fermentation was carried out. Fifty-eight samples from the digesters were collected at different time points, referred as two anaerobic digesters influent (S0 and W0) and 56 anaerobic digesters effluent samples (S4 to S60 and W4 to W60).
Influent (S0 and W0) and effluent (S4 to S60 and W4 to W60) samples (20 mL) were collected using sterile bottles. All samples were sent to the laboratory and stored at 4 °C. Volumes of 10–15 mL of each sample were subjected to sonication on ice for 1 min (cycle, 1.0; amplitude, 100 %) with an ultrasonic homogeniser (Vibra Cell VCX130PB; Sonics & Materials, USA). Aliquots of 5 mL of each sample were centrifuged for 15 min at 14,000g and 4 °C, and the supernatants were removed. The genomic DNA was then extracted using the QIAamp DNA stool mini kit (QIAGEN, Heidelberg, Germany) according to the manufacturer’s instructions. The DNA concentrations were quantified using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA), whereas its integrity was visually examined by gel electrophoresis on a 0.8 % (w/v) agarose gel stained with ethidium bromide. The extracted DNA was stored at −20 °C until use.
Amplification of the V3 region of the 16S rRNA Gene
To analyse the dynamics of the bacterial and archaeal communities, the V3 region of the 16S rRNA gene was amplified using the specific primers W49F-W104R for Bacteria and W274R-W275F for Archaea (Table S1) according to Braun et al. [13]. PCR amplifications were carried out using Mastercycler thermocycler (Eppendorf, Hamburg, Germany). Amplification of samples was checked using an Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Palo Alto, USA).
CE-SSCP DNA Fingerprinting
For CE-SSCP electrophoresis, products were diluted in water (1/10) and added to an SSCP mix with 18.925-μL formamide (Genescan-Applied Biosystems, Norwalk, CT, USA) and 0.075 μL internal standard (ROX, Genescan-Applied Biosystems). The mixture was denatured at 95 °C for 5 min and immediately cooled in ice for 10 min. CE-SSCP electrophoresis was carried out in an ABI Prism 3130 genetic analyser (Applied Biosystems, Foster City, CA, USA) with capillary tubes (47 × 50 μm) filled with 5.6 % non-denaturing conformation analysis polymer (Applied Biosystems). CE-SSCP analysis provides a pattern of peaks and these data were analysed using GeneScan software (Applied Biosystems) and the StatFingerprints R library [14].
Quantitative PCR
Quantitative PCR reactions (qPCR) were prepared using 96-well real-time PCR plates (Eppendorf, Hamburg, Germany) and a Mastercycler ep Gradient S (Eppendorf, Hamburg, Germany). For the quantification of the bacterial 16S rRNA gene, the primers used were W208 (250 nM) and W209 (250 nM) and probe was W210 (50 nM). For Archaea, the 16S rRNA gene primers were W211 (250 nM) and W212 (250 nM) and the probe was W213 (50 nM), according to Yu et al. [15] (Table S1). The quantitative amplification reactions were carried out in a total volume of 25 μL. Thus, all reaction mixtures contained 12.5 μL of Express qPCR Supermix with premixed ROX (Invitrogen, France), 5 μL of template DNA with two appropriate dilutions (0.1 and 1 ng μl−1), 10 nM forward and reverse primers, and 8 nM TaqMan probe, with water added to obtain a final volume. The following thermal programme was used: initial incubation of 20 s at 95 °C and 40 cycles of denaturation (95 °C, 15 s; 60 °C, 1 min). Samples were run in triplicate. Results were compared with a standard curve to obtain the number of target copies in the sample. Standard curves were generated for each assay by using 10-fold dilutions of pEX-A plasmids (Eurofins MWG Operon, Ebersberg Germany) containing 16S rRNA sequence of Escherichia coli pk3 (X80731) and 16S rRNA sequence of Methanobrevibacter smithii (U55233). The total number of Bacteria or Archaea was finally expressed as the number of target copies per gramme of effluent or influent.
Sequencing of 16S rRNA Gene
DNA extracts from the samples collected after 60 days of fermentation were selected for high-throughput sequencing analysis. A mixture of amplicons generated from F515 and R909 primers (Table S1) was sequenced on a 454 GS-FLX Titanium sequencer (Roche Life Sciences, USA) by the Molecular Research Laboratory (TX, USA) based upon established and validated protocols [16]. These primers region were selected in order to target both Bacteria and Archaea present in the samples.
After sequencing, all failed sequence reads, low quality sequence ends and tags were removed. Mothur was used to remove the redundancy of the tag sequences and select unique tag sequences (each unique tag therefore represented different numbers of tag sequences). Briefly, flowgrams were quality controlled and de-noised with the trim.flows, shhh.flows, trim.seqs, unique.seqs commands, aligned against the Silva reference database using align.seqs, and chimera-checked with the chimera.uchime command. Using the classify.seqs command, the final sequence dataset was classified considering the Silva taxonomy distributed with Mothur. The number of sequences obtained was 17,962 from all samples. Operational taxonomic units (OTUs) were defined as group of sequences with a p distance of 3 % (or 97 % similarity), resulting in the successful construction of a distance matrix by the commands dist.seqs and cluster. OTUs were then taxonomically classified using BLASTn against the Silva reference database.
To characterise the level of similarity within each library in both seasonal conditions, the most prevalent Bacteria and Archaea OTUs were represented by calculating the percentage of the total population that they accounted for. Shannon-Weaver (Hʹ) and Simpson diversity indices (1-D) with 95 % confidence intervals were used to assess the diversity present in samples.
Nucleotide sequence accession numbers
Sequence data associated with this study were deposited in the GenBank Short Read Archive (SRA) under the accession number SRP041213.
Data Analysis
To report an average performance in two ambient temperature anaerobic processes (summer and winter), the two different bioreactors were treated as replicates. Average and standard errors were determined.
In order to investigate whether bacterial and archaeal communities differed between the two ambient temperature anaerobic digestion conditions, the ratio of Archaea to Bacteria was estimated by calculating the average numbers of archaeal and bacterial cells. The average numbers of archaeal and bacterial cells were estimated using all samples pendant digestion.
Differences in bacterial and archaeal quantity among different seasons were determined with an analysis of variance (ANOVA) procedure for independent samples to test for statistically significant differences. The ANOVA test was used to determine whether there were significant differences between groups’ density and seasons, to compare production of methane and to study the differences in the Archaea/Bacteria ratios. A p value of p < 0.05 was considered to indicate significance.
All SSCP data analyses were done in the software environment R. Community data as CE-SSCP profiles were aligned and normalised with the package StatFingerprints. Variation seasonal of bacterial and archaeal communities’ data were visualised using principal component analysis (PCA). Principal component analysis is a method used to reduce the number of dimensions of a multivariate dataset like environmental data. The matrices of similarity were calculated on the basis of Euclidean distances between each pair of CE-SSCP profiles. The Simpson diversity index (D) was also computed for each fingerprinting profile using the implementation of the R StatFingerprints library.
Results
Physicochemical Characteristics of the Pilot-Scale Bioreactors
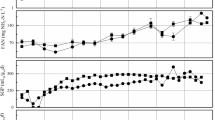
Over the 60 days of hydraulic retention time, two ambient temperature pilot-scale anaerobic reactors treating dairy cattle manure were sampled in different seasons. Ambient temperature was in the mesophilic range, between 14 and 25 °C in winter and 24 and 35 °C in summer. The pH was between 6.95 and 7.41 during the entire study. In all digesters, reductions in total and volatile solids were observed, the reduction rate in TS of fermenting cattle manure was about 65 % and VS was 68 %. Total chemical oxygen demand in the influent was 55.50 (±4.2) g L−1 (summer) and 44.68 (±3.6) g L−1 (winter), approximately 31 % of which was present in the form of dissolved chemical oxygen demand. The average daily biogas produced in the summer and winter months was 18.7 (±9.17) and 16 (±6.2) L day−1, respectively, these differences were significant (p < 0.0001). This indicated that lowering of the temperature from 29.5 to 19.5 °C resulted in a 14.5 % reduction in daily biogas production. There was no significant difference in the methane content of the biogas produced in summer (59.2 %) and winter (53.7 %) months (ANOVA, p = 0.73). There was no significant difference in the average methane yield between the reactors operated in the summer and winter months, as shown in Fig. 2 (ANOVA, p = 0.60).

Methane yield from dairy cattle manure as functions of the seasonal (summer and winter) and temporal patterns (during 60 days of fermentation). The data are expressed as the mean (n = 2)
Overview of Microbial Community
Abundance Overtime of Bacteria and Archaea
The influence of seasonal patterns (summer and winter) and temporal patterns (during 60 days of fermentation) on the number of Bacteria and Archaea was determined by qPCR in all samples (n = 58). All results were similar from each replica, in both digesters operated in the summer (ANOVA, p = 0.999) and in the two operated in the winter (ANOVA, p = 0.809). For each sample during anaerobic processes, the concentration of bacterial and archaeal V3 gene copies per gramme of effluent or influent was assessed using the average of the two replicas operated during each season (Fig. 3). The number of 16S rRNA gene copies was 2.8 × 109 and 1.4 × 108 for Bacteria and 6.4 × 109 and 2.2 × 108 for Archaea in the summer and winter samples, respectively. No significant differences in abundances were found for bacterial communities (ANOVA, p = 0.700) and the total number of Archaea (ANOVA, p = 0.959) in different seasons, suggesting that both communities, independent of the season, obtained a similar dynamics and abundance during the 60 days of fermentation.

Plot of absolute abundance of Bacteria and Archaea by qPCR as functions of the seasonal (summer and winter) and temporal patterns (during 60 days of fermentation). All Bacteria and Archaea data are expressed as log10 copies per gramme of samples. Values indicate mean (n = 10) and the x-axis represents the time (days) of sampling
The Archaea/Bacteria ratio was also studied using qPCR data. The ratio varied between 1 and 11 % (mean 6 %) in the summer samples and 1 and 9 % (mean 4.5 %) in the winter. The ANOVA test revealed no significant difference (p = 0.277), indicating that no clear shift occurred during the 60 days of fermentation between the Archaea/Bacteria ratios in both summer and winter months.
Dynamics of Bacteria and Archaea Communities
The dynamic temporal and seasonal results of the bacterial and archaeal communities were obtained by CE-SSCP analysis and their evolution was examined during both seasons and during anaerobic processes. The resulting microbial community profiles were aligned and compared in order to highlight the genetic distances between the samples. Euclidean distances were computed and the evolution of both communities is shown using principal components analysis (PCA) (Fig. 4a). Thus, PCA plots the dissimilarity in the bacterial community dynamics between the 58 samples and close to 50 % of the variation was explained by PC1 and PC2.

a Principal component analysis plot of SSCP profiles between bacterial communities data and replicate samples (a and b) collected at different times (S summer and W winter). Labels (0–60) indicate the time (days) of sampling. Percentages on axes are per cent of total variation in dataset explained. b SSCP patterns from S60a and S60b (summer effluent samples) and W60a and W60b (winter effluent samples)
Two distinct stages were observed for the initial samples in the summer (S0 to S11) and winter (W0 to W8) samples, suggesting that the start-up or initial stages are differentiated for two seasons. After this, the consecutive time points showed higher similarity, but no differences were observed between the seasons, probably because all reactors progressed towards a steady state.
The PCA plot revealed that the bacterial community structure was influenced intensively by temporal and seasonal conditions. The Simpsons diversity index between seasonal (summer and winter) and replicas (a and b) samples were close to 0.9 for all of the reactors, showing that the dynamics of the bacterial communities was stable during both process.
The data from the CE-SSCP profiles of final samples are presented in Fig. 4b. The relative diversity of the bacterial community was represented by the number of peaks on the profiles. Overall, these profiles presented complex communities structures. As shown in Fig. 4b, the individual profiles were highly similar under same seasons, suggesting a high reproducibility of the reactors.
The archaeal community structure was less complex than the bacterial community structure. Regarding the Archaea patterns, over the time points, comparatively similar community structures were observed in summer and winter samples. The CE-SSCP profile obtained showed a low diversity with the mean Simpson diversity index between 0.8 and 0.9 for both seasons (data not shown).
Microbial Community Structures
Final samples from summer (S60a and S60b) and winter (W60a and W60b) ambient temperature pilot-scale bioreactors were subjected to microbial diversity analysis using the 16S rRNA gene. The relative abundance of archaeal and bacterial groups in the different anaerobic reactors was analysed and the percent of sequences belonging to specific taxonomic groups are summarised in Fig. 5. All sequences in the summer (S60a and S60b) and winter (W60a and W60b) samples were represented by 15 phyla. The average value for the rate Bacteria accounted for 97.8 % of the sequences detected in the reactors, while the rest belonged to Archaea (Table 1).

Phylogenetic distribution of the bacterial and archaeal sequences retrieved from S60a and S60b (summer effluent samples after 60 days of fermentation) and W60a and W60b (winter effluent samples after 60 days of fermentation). a and b indicate replicate samples
Bacterial Community Composition
The sequence analyses at the taxonomical bacterial identified 13 phyla and 17 classes in all reactors. All percentages given below represent the relative abundance of the bacterial sequences detected. The phylum Firmicutes (low-G+C, Gram-positive bacteria) was the most abundant in all bioreactors, representing 51.9–57.2 % of the summer sequences (S60a and S60b, respectively) and 48.0–45.5 % of the winter (W60a and W60b, respectively). Within the Firmicutes, sequences belonging to the Clostridia class dominated (97.3 % in the summer samples and 95.0 % in the winter of total Firmicutes sequences).
Sequences belonging to Bacteroidetes were the second major phylum, constituting 35.1–27.8 % of S60a and S60b, respectively, and 42.7–46.2 % of W60a and W60b, respectively. The predominant family was Bacteroidaceae, with around 90.0 % of the Bacteroidetes sequences, and Sphingobacteria with an average of 5 % for all samples.
Planctomycetes was the next most common phylum, representing 3.1–4.1 % of S60a and S60b, respectively, and 1.6 % of the winter samples, followed by Actinobacteria (up to 1.1 %) and Spirochetes (up to 1 %). Different classes of Proteobacteria were found in both conditions in quite small numbers and were represented mainly by γ-proteobacteria and δ-proteobacteria, while β-, α-proteobacteria contributed <1 % of the total number of sequences in all reactors.
Furthermore, sequences belonging to Caldiserica, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Lentisphaerae, Spirochaetes, Synergistetes and Verrucomicrobia were identified and accounted on average for 1 % of the total of each sample.
Archaeal Community Composition
Phylogenetic affiliations showed that Archaea have only two phyla identified: Euryarchaeota and Crenarchaeota. All percentages mentioned below reflect the relative abundance of the archaeal sequences detected.
All of the reactors were dominated by sequences from the phylum Euryarchaeota accounted for 99.4–93.0 % of the summer sequences (S60a and S60b, respectively) and 100.0–97.1 % of the winter (W60a and W60b, respectively), while the rest belonged to Crenarchaeota.
Within the phylum Euryarchaeota, sequences belonging to the Methanobacteriaceae family dominated in all reactors, 53.4–69.0 % of S60a and S60b, respectively, and 35.7–40.5 % of W60a and W60b, respectively, in which the hydrogenotrophic Methanobrevibacter sp. accounted for the majority of the sequences (26.6 % in S60a; 43.4 % in S60b; 37.9 % in W60a; 25.7 % in W60b).
Other detected phylum of Euryarchaeota sequences were assigned to the Methanosarcinales (4.2–6.9 % of S60a and S60b and 27.8–20.0 % of W60a and W60b, respectively) and Methanomicrobiaceae family, in which the Methanocorpusculum sp. accounted for the majority of sequences.
Microbial Community Structures at OTU Level
In order to compare the microbial structure among these samples, operational taxonomic units (OTUs) were determined for each sample at distance levels of 3 % (Table 1). In total, 17,531 sequences were classified into 1710 OTU (1676 OTU for the domain Bacteria; 34 OTU for the Archaea).
Among the 20 most prevalent OTUs shown in Table 2, it was possible to visualise 36.8 to 49.2 % of the bacterial structure and in the top ten dominant among Archaea OTUs (Table 3), it was possible to evaluate the distribution from 79.7 to 89.1 % of all archaeal sequences. These results are consistent with the phylum assignment data. For example, 10 of the 20 most prominent Bacteria OTUs from the summer and winter sequences are members of the phylum Firmicutes (the most predominant phylum), nine are from the phylum Bacteroidetes (the second largest phylum), and one is from the phylum Planctomycetes (the third most predominant phylum).
The 20 most prominent OTUs were affiliated with mainly uncultured bacteria from different anaerobic habitats (rumen and anaerobic digester) and only remotely related to known bacterial species. Only one small group (B6, B9 and B14) showed sequence identity less than 98 % similarity to sequences in the public database.
The breakdown of the 10 most prominent Archaea OTU is presented in Table 3. All belonged to the phylum Euryarchaeota, and most of the archaeal sequences were related to sequences of known archaeal species from several different environments. Only A7 of the 10 OTU showed sequence identity less than 99 % similarity to sequences in the public database. The most dominant OTU (A1 and A2) were affiliated with microorganisms in the family Methanobacteriaceae, corresponding to 68–70 % of the S60a and S60b sequences and 48–60 % of W60a and W60b.
The bacterial and archaeal phylotype dynamic levels were observed using Simpson and Shannon diversity indices. Simpson and Shannon indices (Table 1) indicated approximately equal diversity in both seasons. These results indicate a higher diversity of bacterial communities compared to Archaea. All results were reproducible from each replica (a and b).
Discussion
In this study, we present an overview of the microbial community dynamics and diversity regarding bacteria and archaea residing in pilot-scale anaerobic digestion, operating at ambient temperatures in Brazil, by using different methodological approaches. The dynamics of microbial community changes was investigated by molecular fingerprinting (CE-SSCP) to evaluate the temporal variation in the community structure along the anaerobic digestion process, and by quantitative PCR techniques to evaluate abundance variation between the two prokaryotic domains. To study microbial diversity, molecular fingerprinting (SSCP) was employed to qualitatively investigate the distribution of bacteria and archaea according to the temporal and seasonal parameters, and to investigate bacteria and archaea species distribution at the end of the process, massive sequencing was performed.
Analysis of the temporal bacterial and archaeal dynamics (during 60 days of fermentation) revealed that both summer and winter anaerobic digestion processes contain similar microbial abundance, and that the slight temperature variation between seasons was not definitive for a wide dynamics variation between the digesters. The similar concentrations of bacterial and archaeal 16S rRNA gene copies per gramme of samples (Fig. 3) and values of the Simpson index during the summer compared to the winter process indicated an equal microbial dynamics and abundance over the course of the fermentation time in both seasons. In this study, values of absolute abundance of Bacteria and Archaea are in agreement with other community analyses in anaerobic reactors [17].
Even with the results of quantitative PCR, anaerobic digestion process showed no difference in Archaea/Bacteria ratios between the two seasons. Thus, different conditions seem to yield large variations in archaeal/bacterial ratios, and our results were in the same order as the values reported in the literature. Archaeal/bacterial ratios have been previously reported at 0–7 % in a full-scale biogas reactor fed with animal manure (pig and cow) [5], 6 % in a reactor fed with fodder beet sludge [18], and 0.5–4.5 % in anaerobic digesters fed with liquid manure and garden refuse [19]. Furthermore, the abundance of bacteria was higher than that of Archaea, corroborating data from other studies [20, 21].
Analysis of CE-SSCP profiles was employed to address temporal variation of the bacterial and archaeal community composition and to analyse the structural variation between different seasons or the dynamics of the same system. The results showed that the dynamics among the seasons were similar (Fig. 4a). Thus, samples of both seasons shifted in the same pattern. This indicated that the initial microbial communities had the capability of adaptation and that the community succession occurred at similar times for both of the seasons evaluated. Time of fermentation and factors such as digester operational conditions and digester performance had a great effect on microbial dynamics. Other studies also showed a shifting community in the initial stages of the mesophilic process with a tendency towards stabilisation after a period of fermentation in animal manure [8, 22, 23]. The replicated ambient temperature reactors (a and b) showed equal performance and were highly comparable.
Regarding the biogas production, as for the similarity in community dynamics and abundance, both seasons’ reactors appeared similar concerning performance, i.e., there were equal volatile solid reductions and methane yield. Data for all reactors showed stable anaerobic digestion in the summer and winter months. Few researches have been identified regarding the ambient temperature of anaerobic digestion in the mesophilic range of animal manure. It has been shown that methane production from animal manure follows the same pattern at mesophilic (35 °C) and ambient (16.8–29.5 °C) temperatures [24]. Thus, the methane yield obtained was similar to other studies of mesophilic biogas production from animal manure [25–27].
Phylogenetic affiliation showed that the anaerobic process for both seasons had a feature in common: the dominant composition of two bacterial phyla in the final samples (Fig. 5). However, it was possible to observe small changes in percentage between the phyla identified. The Shannon Index was used to characterise diversity in the same final data and the values for the Bacteria and Archaea group revealed slight variability in the summer months. However, based on the CE-SSCP peaks, a slight change in the structure of Bacteria and Archaea was detected (Fig. 4b). Thus, even though our results showed the constant abundance and dynamics of communities during the digestion process, a subtle bacterial and archaeal structural change was observed. The diversity and composition of the microbial community varied with ambient temperature along the anaerobic digestion process, probably due to seasonal changes and temporal patterns.
The most prevalent bacterial communities were Firmicutes and Bacteroidetes, which are responsible for complex organic matter degradation and fermentation. The dominance of these phyla in biogas reactors is in agreement with other community analyses in anaerobic bioreactors [21, 23, 28, 29]. In this study, within the phylum Firmicutes, class Clostridia dominated in the bacterial community. This agrees with the results from other authors, who suggested that many Clostridia are involved in the breakdown of polysaccharides and therefore play an important role in the hydrolysis step of plant biomass [5, 30–32].
The phyla that are less abundant, in particular, Actinobacteria, Proteobacteria, Lentisphaerae, Synergistetes and Verrucomicrobia, are normally found in a wide variety of environments and play various roles in the anaerobic digestion of animal manure [7, 32]. These phyla showed that the percentage of total values differentiated between the seasons, indicating that there are more complex microbial components residing within this ambient temperature system.
In the present study, the number and distribution of OTUs and therefore the methanogen diversity varied slightly between the two seasons. It can be suggested that there are few different pathways for methane formation in the different seasonal reactors based on the results of the composition and structure of the archaeal community shown in this study. The phylum Euryarchaeota was the major methanogenic archaeal group in both seasons. However, the hydrogenotrophic Methanobacteriaceae dominated the archaeal communities in the summer months, while, in the winter months, the dominance was divided between Methanobacteriaceae and Methanosarcinaceae. Beyond changing the microbial structure, temperature variation also caused a small decrease (2.7 L day−1) in the production of biogas at low ambient temperature.
Earlier studies on archaeal populations in mesophilic conditions did not suggest any clear relationship between specific genera and process temperature [5, 7, 20]. However, these results indicate that hydrogenotrophic order would be the key methanogenic group in the ambient temperature bioreactors and that the Methanobacteriaceae community composition might be affected by slight temperature change.
Among Methanobacteriaceae, the A1 and A2 OTU were closely related to species of the genera Methanobrevibacter sp. and Methanocorpusculum sp.; these strains are usually present in the animal rumen and are related to an important role in methane production in environments where there are seasonal variations in temperature. Moreover, the genomes of these genera are significantly enriched with genes involved in the utilisation of CO2, H2 and formate during methanogenesis [33]. Thus, a high percentage of this genus in both seasons, in this study, showed the essential role played by these species in methanogenesis and suggest that this might be linked to the addition of cattle manure in the substrate for these reactors.
The abundance of clones related to the family Methanosarcinaceae has been widely reported earlier in a variety of anaerobic digesters [7, 18]. Many methanogens of the Methanosarcina genus display diverse metabolic features, including the metabolism of both hydrogen and acetate as an energy source [34]. In our study, A3 and A5 OTUs were more prominent in the winter samples library.
Archaeal and Bacterial sequences with a high similarity to the OTU clustered with the uncultured strains from ambient temperature anaerobic processes were described for a wide range of natural environments and artificial systems in different parts of the world, such as in the intestinal tract of animals, wastewater and digesters. Thus, the microbial community seemed to be representative for a biomass degrading anaerobic microbial consortium [18].
Conclusions
Altogether, these results revealed first insights into the slight variation of microbial structure in the ambient temperature biogas process. Setting the optimum digester temperature is one of the most critical factors for economically viable digester operation in tropical countries, such as Brazil, since most annual temperatures are in the mesophilic range. These results show that even with the narrow range of temperature changes, the structure of the microbial community during the fermentation process has undergone a slight change. However, this structure change has not been able to influence the quality of methane production and dynamics communities, leading to the conclusion that this probably occurred due to redundancy of groups, as the communities significantly differed in composition; however, all were able to carry out similar roles.
References
Guerci M, Knudsen MT, Bava L, Zucali M, Schönbach P, Kristensen T (2013) Parameters affecting the environmental impact of a range of dairy farming systems in Denmark, Germany and Italy. J Clean Prod 54:133–141
Abubaker J, Risberg K, Pell M (2012) Biogas residues as fertilisers—effects on wheat growth and soil microbial activities. Appl Energy 99:126–134
Holm-Nielsen JB, Seadi TA, Oleskowicz-Popiel P (2009) The future of anaerobic digestion and biogas utilization. Bioresour Technol 100:5478–5484
Kumar KV, Sridevi V, Rani K, Sakunthala M, Kumar CS (2013) A review on production of biogas, fundamentals, applications & its recent enhancing techniques. Elixir Chem Engg 57:14073–14079
Sundberg C, Al-Soud WA, Larsson M, Alm E, Yekta SS, Svensson BH, Sørensen SJ, Karlsson A (2013) 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full-scale biogas digesters. FEMS Microbiol Ecol 85:612–626
Kunz A, Steinmetz RLR, Rammeb MA, Coldebella A (2009) Effect of storage time on swine manure solid separation efficiency by screening. Bioresour Technol 100:1815–1818
Levén L, Eriksson ARB, Schnurer A (2007) Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic house holdwaste. FEMS Microbiol Ecol 59:683–693
Lee SH, Kang HJ, Lee YH, Lee TJ, Han K, Choic Y, Park HD (2012) Monitoring bacterial community structure and variability in time scale in full-scale anaerobic digesters. J Environ Monit 14:1893–1905
Ferrer I, Gamiz M, Almeida M, Ruiz A (2009) Pilot project of biogas production from pig manure and urine mixture at ambient temperature in Ventanilla (Lima, Peru). Waste Manag 29:168–173
Yusuf MOL, Debora A, Ogheneruona DE (2011) Ambient temperature kinetic assessment of biogas production from co-digestion of horse and cow dung. Res Agr Eng 57:97–104
Nikolaeva S, Sánchez E, Borja R (2013) Dairy wastewater treatment by anaerobic fixed bed reactors from laboratory to pilot-scale plant: a case study in Costa Rica operating at ambient temperature. Int J Environ Res 7:759–766
American Public Health Association (APHA) (2005) Standard methods for examination of water and wastewater, 21st edn. American Water Works Association, Washington, DC
Braun F, Hamelin J, Gévaudan G, Patureau D (2011) Development and application of an enzymatic and cell flotation treatment for the recovery of viable microbial cells from environmental matrices such as anaerobic sludge. Appl Environ Microbiol 77:8487–8493
Michelland RJ, Dejean S, Combes S, Fortun-Lamothe L, Cauquil L (2009) StatFingerprints: a friendly graphical interface program for processing and analysis of microbial fingerprint profiles. Mol Ecol Resour 9:1359–1363
Yu Y, Lee C, Kim J, Hwang S (2005) Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol Bioeng 89:670–679
Tamaki H, Wright CL, Li X, Lin Q, Hwang C, Wang S, Thimmapuram J, Kamagata Y, Liu W (2011) Analysis of 16S rRNA amplicon sequencing options on the Roche/454 next-generation titanium sequencing platform. PLoS ONE 6:e25263
Alexander TW, Yanke JL, Reuter T, Topp E, Read RR, Selinger BL, McAllister TA (2011) Longitudinal characterization of antimicrobial resistance genes in feces shed from cattle fed different subtherapeutic antibiotics. BMC Microbiol 11:19
Klocke M, Mähnert P, Mundt K, Souidi K, Linke B (2007) Microbial community analysis of a biogas-producing completely stirred tank reactor fed continuously with fodder beet silage as mono-substrate. Syst Appl Microbiol 30:139–151
Moletta-Denat M, Bru-Adan V, Delgenès JP, Hamelin J, Wéry N, Godon JJ (2010) Selective microbial aerosolization in biogas demonstrated by quantitative PCR. Bioresour Technol 101:7252–7257
Hori T, Haruta S, Ueno Y, Ishii M, Igarashi Y (2006) Dynamic transition of a methanogenic population in response to the concentration of volatile fatty acids in a thermophilic anaerobic digester. Appl Environ Microbiol 72:1623–1630
Liu FH, Wang SB, Zhang JS, Zhang J, Yan X, Zhou HK, Zhao GP, Zhou ZH (2009) The structure of the bacterial and archaeal community in a biogas digester as revealed by denaturing gradient gel electrophoresis and 16S rDNA sequencing analysis. J Appl Microbiol 106:952–966
Peu P, Brugère H, Pourcher AM, Kérourédan M, Godon JJ, Delgenès JP, Dabert P (2006) Dynamics of a pig slurry microbial community during anaerobic storage and management. Appl Environ Microbiol 72:3578–3585
Bengelsdorf FR, Gerischer U, Langer S, Zak M, Kazda M (2013) Stability of a biogas-producing bacterial, archaeal and fungal community degrading food residues. FEMS Microbiol Ecol 84:201–212
Alvarez R, Villca S, Lidén G (2006) Biogas production from llama and cow manure at high altitude. Biomass Bioenerg 30:66–75
Hill DT, Taylor SE, Grift TE (2001) Simulation of low temperature anaerobic digestion of dairy and swine manure. Bioresour Technol 78:127–131
Rico JL, García H, Rico C, Tejero I (2007) Characterization of solid and liquid fractions of dairy manure with regard to their component distribution and methane production. Bioresour Technol 98:971–979
Chae KJ, Jang A, Yim SK, Kim IS (2008) The effects of digestion temperature and temperature shock on the biogas yields from the mesophilic anaerobic digestion of swine manure. Bioresour Technol 99:1–6
McGarvey JA, Miller WG, Zhang R, Ma Y, Mitloehner F (2007) Bacterial population dynamics in dairy waste during aerobic and anaerobic treatment and subsequent storage. Appl Environ Microbiol 73:193–202
Kampmann K, Ratering S, Kramer I, Schmidt M, Zerr W, Schnella S (2012) Unexpected stability of Bacteroidetes and Firmicutes communities in laboratory biogas reactors fed with different defined substrates. Appl Environ Microbiol 78:2106–2119
Krause L, Diaz NN, Edwards RA, Gartemann KH, Krömeke H, Neuweger H, Pühler A, Runte KJ, Schlüter A, Stoye J, Szczepanowski R, Tauch A, Goesmann A (2008) Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J Biotechnol 136:91–101
Jaenicke S, Ander C, Bekel T, Bisdorf R, Dröge M (2011) Comparative and joint analysis of two metagenomic datasets from a biogas fermenter obtained by 454-pyrosequencing. PLoS ONE 6:e14519
Ritari J, Koskinen K, Hultman J, Kurola JM, Kymäläinen M, Romantschuk M, Paulin L, Auvinen P (2012) Molecular analysis of meso- and thermophilic microbiota associated with anaerobic biowaste degradation. BMC Microbiol 12:121
Simankova MV, Kotsyurbenko OR, Luedersb T, Nozhevnikova AN, Wagner B, Conrad R, Friedrich MW (2003) Isolation and characterization of new strains of methanogens from cold terrestrial habitats. Syst Appl Microbiol 26:312–318
Ma J, Zhao B, Frear C, Zhao Q, Yu L, Li X, Chen S (2013) Methanosarcina domination in anaerobic sequencing batch reactor at short hydraulic retention time. Bioresource Technol 137:41–50
Wang Y, Qian PY (2009) Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PloS ONE 4:e7401
Brosius J, Dull TJ, Sleeter DD, Noller HF (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J Mol Biol 148:107–127
Acknowledgments
The authors are grateful to Kim Milferstedt, for support to the program database analysis. This study was supported by grants from EMBRAPA’s virtual Laboratory abroad (Labex Program) in Europe, Brazilian National Council for Scientific and Technological Development (CNPq), Research Support Foundation of Minas Gerais (FAPEMIG). The authors gratefully acknowledge Embrapa Dairy Cattle, Institut National de la Recherche Agronomique (INRA) and Federal University of Juiz de Fora (UFJF). We also thank Coordination of Improvement of the Personnel of Higher Education (CAPES) by the scholarship (Process 11853-12-3).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 15 kb)
Rights and permissions
About this article

Cite this article
Resende, J.A., Godon, JJ., Bonnafous, A. et al. Seasonal Variation on Microbial Community and Methane Production during Anaerobic Digestion of Cattle Manure in Brazil. Microb Ecol 71, 735–746 (2016). https://doi.org/10.1007/s00248-015-0647-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-015-0647-y