
Abstract
Gut microbiota is increasingly regarded as an integral component of the host, due to important roles in the modulation of the immune system, the proliferation of the intestinal epithelium and the regulation of the dietary energy intake. Understanding the factors that influence the composition of these microbial communities is essential to health management, and the application to aquatic animals still requires basic investigation. In this study, we compared the bacterial communities harboured in the intestines and in the rearing water of grass carp (Ctenopharyngodon idellus), crucian carp (Carassius cuvieri), and bighead carp (Hypophthalmichthys nobilis), by using 454-pyrosequencing with barcoded primers targeting the V4 to V5 regions of the bacterial 16S rRNA gene. The specimens of the three species were cohabiting in the same pond. Between 6,218 and 10,220 effective sequences were read from each sample, resulting in a total of 110,398 sequences for 13 samples from gut microbiota and pond water. In general, the microbial communities of the three carps were dominated by Fusobacteria, Firmicutes, Proteobacteria and Bacteroidetes, but the abundance of each phylum was significantly different between species. At the genus level, the overwhelming group was Cetobacterium (97.29 ± 0.46 %) in crucian carp, while its abundance averaged c. 40 and 60 % of the sequences read in the other two species. There was higher microbial diversity in the gut of filter-feeding bighead carp than the gut of the two other species, with grazing feeding habits. The composition of intestine microbiota of grass carp and crucian carp shared higher similarity when compared with bighead carp. The principal coordinates analysis (PCoA) with the weighted UniFrac distance and the heatmap analysis suggested that gut microbiota was not a simple reflection of the microbial community in the local habitat but resulted from species-specific selective pressures, possibly dependent on behavioural, immune and metabolic characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In vertebrates, the gastrointestinal tract harbours a complex ecosystem that is colonised by a large, diverse and dynamic collection of microorganisms [1]. After millions of years of co-evolution with the host, the gut microbiota may be considered as an integral component of the host. This microbial community plays a key role in the maintenance of normal gut function, including the defence against pathogens, the proliferation of enterocytes, the digestion of complex carbohydrates and the production of secondary metabolites, such as vitamins [2–4]. In addition, the gut microbiota may harbour opportunistic pathogens, indicating that the gastrointestinal tract is a potential pathway for pathogen invasion [5, 6]. Consequently, the intestinal microbiota should be further investigated in farmed fish, due to the importance of health management in aquaculture.
The composition of gut microbial communities is affected by exogenous and endogenous factors in fish, including developmental stage, gut structure, diet composition and trophic level, habitat and surrounding environment (e.g. water temperature and salinity), rearing conditions and possibly, phylogenetic position [7, 8].
The gut microbiota have been analysed intensely by using both culture-dependent and culture-independent methods. Many microbes found in the gastrointestinal tract were not identified, due to the difficulty of culturing them routinely in the laboratory [9–11]. Culture-independent molecular techniques like denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (T-RFLP) and PCR cloning and sequencing have been currently applied, and this revolutionised the perception of the microbial communities [5, 6, 12–14]. However, only the predominant microorganisms were identified by using these methods, leaving in the dark most of the microbial diversity [15]. High-throughput sequencing has made accessible the in-depth analysis of microbial communities. Sequence tags can be used to enable the simultaneous processing of large numbers of samples with limited costs [16]. Several recent studies described the exploitation of these technologies that mainly focused on gut microbiota in humans [17] and other terrestrial vertebrates [18]. The method is increasingly applied to fish microbiota [5, 12, 19].
Grass carp (Ctenopharyngodon idellus), crucian carp (Carassius cuvieri) and bighead carp (Hypophthalmichthys nobilis) are the major carp species in Chinese aquaculture and widely cultivated for food. The production of these species of fish in China reached 9.41 million tons (bighead carp, 2.67 million tons; grass carp, 4.44 million tons; and crucian carp, 2.30 million tons) in 2011, accounting for about 43.05 % of the total freshwater-cultured fish annual output [20]. The polyculture of carps with different feeding habits is a traditional method to optimise the use of trophic resources in ponds [21]. In order to improve the health and productivity of these important aquaculture species, it is essential to characterise their gut bacterial communities and the factors that influence the composition and stability of the microbiota.
Thus far, several studies have shown differences in gut microbiota composition between fish species. These differences were mainly detected at the phylum level, relying on molecular-based fingerprinting methods (e.g. DGGE and TTGE) [13, 14]. The principal aim of this study was (1) to investigate and to compare the gut microbiota composition in the three species of fish that cohabitated in the same environment by utilising high-throughput sequencing technology and (2) to try to relate the composition of the gut microbial communities to the biological characteristics of the three species.
Materials and Methods
Sample Collection
The adult grass carp, crucian carp and bighead carp were sampled from the same earth pond with an approximate size of 0.5 ha, which was located at Dongxihu Fish Farm, Wuhan (mid-China).The fish were fed twice a day with a commercial fish food (crude protein, ≥30.0 %; crude fibre, ≥12.0 %; crude ash, ≤15.0 %; calcium, 0.4–2.5 %; phosphate, ≥0.7 %; salt, 0.3–1.2 %; moisture, ≤12.5 %; and lysine, ≥1.2 %; Wuhan JiuZhou Shennong Pharmaceutical Co., Ltd, China).The fish were sampled on 10th April, 2012, as the water temperature in the pond was 18 °C. Three healthy individuals of each species were randomly harvested with nets. The average body weights were c. 1.2, 0.3 and 1.0 kg for grass carp, crucian carp and bighead carp, respectively. The fish were carried to the laboratory in ice within 2 h. The fish were then euthanized by a high dose of MS 222 (Sigma, Germany).The surface of the fish was rinsed with sterile distilled water, and then with 70 % ethanol to reduce contamination, before dissection with flamed sterile scissors. The intestines were removed aseptically from their abdominal cavity and the content of each fish was squeezed out and separately harvested [12]. The intestinal contents from the three individuals of each species were labelled C1–C3 for grass carp, J1–J3 for crucian carp and Y1–Y3 for bighead carp. Thereafter, the epithelial intestinal mucosa of the three fish from each species was collected and pooled per species to obtain a sufficient bacterial DNA concentration [12, 22]. The mucosal samples were labelled CB, JB and YB for grass carp, crucian carp and bighead carp, respectively. Meanwhile, the pond water was also sampled at an approximate depth of 50 cm from three sites in the pond and pooled together. One litre of water sample was centrifuged at 12,800×g (Thermo) for 20 min at 4 °C, the pellet was collected, labelled Wt and kept frozen at −80 °C until DNA extraction.
DNA Extraction and Purification
The samples (either 250 mg of intestinal content, pooled mucosa or centrifugal pellet from 1 L water) were thawed on ice, and then total bacterial DNA was extracted with the E.Z.N.A. Stool DNA kit (OMEGA, Bio-Tek, USA) according to the manufacturer’s protocol. The integrity of the nucleic acids was determined visually after electrophoresis on 1.2 % agarose gel containing ethidium bromide. The DNA concentration was measured by using a fluorescence spectrophotometer (ES-2, Malcom, Japan). All samples were extracted in duplicates, and the extracts from the same sample were pooled together to avoid bias [6, 12]. The extracted DNA was stored at −80 °C until use.
PCR Amplification and Pyrosequencing
PCR was performed from each sample to produce a fragment of the 16S rRNA gene (~420 bp, covering the V4/V5 hypervariable regions). The universal Bacterial primers 515 F (5′-GTGCCAGCMGCCGCGGTAA-3′; [23]) and 926R (5′-CCGTCAATTYYTTTRAGTTT-3′; [24]) were modified by adding ligation adaptors and/or MID barcodes (454 Life Sciences) to the 5′-ends (Table S1). The PCR was run in triplicates for each sample in a total reaction volume of 20 μL. Each 20 μL reaction mixture contained 1.25 U Takara Ex Taq DNA polymerase, 2 μL 10× Ex Taq buffer (Mg2+), 1.6 μL dNTP mix (all TaKaRa Biotechnology Co., Ltd., Dalian, China), forward and reverse primers (10 pmol each), 1 μL BSA (10 mg mL−1), 1 μL DNA template (~5 ng) and sterile water q.s. ad 20 μL. The PCR conditions were as follows: initial denaturing, 95 °C for 5 min, followed by 30 cycles of 30 s at 94 °C (denaturing), 30 s at 56 °C (annealing) and 30 s at 72 °C (extension) and final extension of the unfinished products for 10 min at 72 °C. The PCR products were migrated on agarose gel and purified by using the QIAEX II Gel Extraction Kit (QIAGEN). Pyrosequencing was performed at the Chinese National Human Genome Center in Shanghai with the 454 GS FLX Sequencing System (Roche).
Statistical and Bioinformatics Analysis
Sequence processing, operational taxonomical units (OTUs)-based approaches, phylotype analysis and hypothesis testing were mainly performed with the software Mothur [25].The sequences were firstly processed using the ‘trim.seqs’ script to minimise the effects of poor sequence quality and sequencing errors. As a result, reads with length <200 bp, average Phred quality score of <20 or mismatches in the tags, were discarded. Sequences with homopolymers longer than 8 bp, any ambiguous bases call and errors in the primer and barcode sequences were removed from the dataset. The pyrosequencing sequences were simplified by using the ‘unique.seqs’ script to generate a unique set of sequences, and then they were aligned by using the ‘align.seqs’ script with the Bacterial SILVA database, which contains 50,000 columns long and which aligned 14,956 bacterial sequences [26]. The aligned sequences were further trimmed, and the redundant sequences were eliminated with the scripts ‘screen.seqs’, ‘filter.seqs’ and ‘unique.seqs’, successively. The resulting sequences after quality control were denoised by using the Mothur-based reimplementation of the PyroNoise algorithm (shhh.flows) with the default parameters [27]. The ‘chimera.slayer’ script was used to identify and remove potentially chimeric sequences. Subsequently, all sequences classified as cyanobacteria/chloroplast were removed to focus on the diversity of bacterial symbionts in guts, as they likely represent contaminants from chloroplast/plant material. The datasets were rarefied in order to equalise sampling effort across samples. In addition, the sampling effort was equalised to the size of the smallest sample (6,218 reads) by random subsampling. Distance matrices were constructed using the ‘dist.seqs’ script and the OTUs were assigned by using the furthest neighbour clustering algorithm (95 to 99 % sequence similarity). OTU rarefaction curves, Good’s coverage and all community richness and diversity indices (ACE, Chao1, Shannon and Simpson) were generated with Mothur [25]. The OTUs defined by a 3 % distance level were phylogenetically classified after the Ribosomal Database Project (RDP) Classifier programme, with a confidence threshold of 50 % [28, 29]. Heatmap figures and Venn diagrams were generated The R Project for Statistical Computing (http://www.r-project.org/). The p test significance analysis and principal coordinates analysis (PCoA) of the Fast UniFrac metric matrix were used to compare the bacterial communities, based on phylogenetic information [30]. S Chao1 and S ACE were calculated for each library according to Kemp and Aller (http://www.aslo.org/lomethods/free/2004/0114a.html) [31]. Because of the non-normal distribution of the samples, the statistical evaluation was performed with nonparametric tests (SPSS, version 19.0). Differences between two independent groups were evaluated by the Mann–Whitney test, and differences between multiple independent groups were evaluated by the Kruskal–Wallis test. The sequences obtained in this study were uploaded and available through the NCBI/EBI/DDBJ Sequence Read Archive (accession number DRA001264).
Results
General Analyses of the Pyrosequencing-Derived Dataset
After filtering the low-quality reads, trimming the longer homopolymer runs, adapters, barcodes and primers, removing all cyanobacteria/chloroplast sequences and rarefying the datasets, 6,218 to 10,220 effective sequences were collected from each sample, resulting in a total of 110,398 sequences from the 13 samples. All the sequences were delineated into OTUs with different sequence similarity values ranging from 95 to 99 %. The number of OTUs from each sample at the various similarity cut-off levels was 1,102 ± 563 (99 % sequence similarity), 708 ± 653 (98 % sequence similarity), 505 ± 396 (97 % sequence similarity), 401 ± 341 (96 % sequence similarity) and 341 ± 299 (95 % sequence similarity). A 97 % sequence identity of the 16S rRNA gene was commonly used to determine the phylotypes at the species level, in accordance with most of the available studies [5, 12, 32].The S Chao1 and S ACE were calculated for each library to assess whether the libraries were large enough to yield a stable estimate of the phylotype richness at the 97 % similarity threshold. We found that the S Chao1 and S ACE richness estimators did not reach an asymptote but tended to converge into a narrow range that exceeded the unstable estimator-producing stage when the subsample sizes approached the actual library sizes (Fig. S1), indicating that these libraries were large enough to yield stable and unbiased estimates of phylotype richness [31, 33]. Therefore, in the rest of this report, a 97 % similarity threshold was used to define OTUs, consistently with the other studies using deep sequencing methods.
A total of 3,897 OTUs were obtained at the 97 % similarity level. Each sample contained 125 to 1,260 OTUs (Table 1). The average read length was 250 bp. The completeness of sampling was estimated with Good's coverage, by calculating the probability that a randomly selected amplicon sequence was already detected in the same sample. The coverage ranged from 89.82 to 99.26 % (96.11 ± 3.37 %), indicating that between 9 and 100 (1/(1 − ‘Good’s coverage’)), additional reads would need to be sequenced before detecting a new phylotype. This level of coverage suggested that the majority of bacterial phylotypes present in the samples were identified in this study. The number of OTUs covered 40–66 and 55–68 % of the richness estimated by the ACE and Chao1 indices, respectively (Table 1). The corresponding rarefaction curves tended to reach the saturation plateau (Fig. S2). The diversity of the microbial community in the intestinal content was significantly different between the three species of fish, as evaluated with the indices of Shannon and Simpson (p < 0.05; Kruskal–Wallis). This diversity was much higher in the filter-feeding bighead carp, compared with the two other grazing species (Table 1).
Composition of Bacterial Community
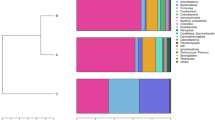
The phylogenetic classification of sequences from the samples resulted in 17 different phyla or groups that were identified in this study. The sequences that could not be classified into any known group were assigned as ‘unclassified bacteria’. The 13 samples showed highly dissimilar 16S rRNA profiles of relative abundance, even at the phylum level (Fig. 1).

Bacterial composition of the different communities (% of relative read abundance of bacterial phyla within each community). Lanes C1–C3, intestinal content of the three individuals of grass carp; lanes J1–J3, crucian carp; and lanes Y1–Y3, bighead carp; CB samples collected from intestinal mucosa and pooled from the three individuals of grass carp, JB samples collected from intestinal mucosa and pooled from the three individuals of crucian carp, YB samples collected from intestinal mucosa and pooled from the three individuals of bighead carp, Wt water sample
The water sample included 13 phyla, where Bacteroidetes (62.91 %), Proteobacteria (26.54 %), Actinobacteria (3.18 %), Fusobacteria (2.28 %) and Verrucomicrobia (0.98 %) were the most abundant groups and accounted for 95.90 % of the reads.
The intestinal content samples were dominated by four major phyla, representing approximately 93.81 % of the sequences, including Fusobacteria (grass carp, 64.58 ± 12.84 %; crucian carp, 97.50 ± 0.40 %; and bighead carp, 41.75 ± 14.62 %), Firmicutes (grass carp, 9.72 ± 6.26 %; crucian carp, 0.42 ± 0.36 %; and bighead carp, 18.93 ± 1.46 %), Proteobacteria (grass carp, 5.95 ± 3.96 %; crucian carp, 1.81 ± 0.39 %; and bighead carp, 16.77 ± 8.09 %) and Bacteroidetes (grass carp, 19.03 ± 6.42 %; crucian carp, 0.11 ± 0.12 %; and bighead carp, 4.85 ± 2.98 %). Actinobacteria were identified in all fish samples (grass carp, 0.09 ± 0.11 %; crucian carp, 0.09 ± 0.02 %; and bighead carp, 0.81 ± 0.14 %). Planctomycetes were only present in one intestinal content sample of crucian carp at 0.10 %, but they were present in all of the samples from grass carp (0.08 ± 0.10 %) and bighead carp (6.88 ± 1.23 %). Similarly, Chloroflexi (3.12 ± 1.74 %), Acidobacteria (3.14 ± 1.60 %) and Verrucomicrobia (1.09 ± 0.34 %) were present in all the intestinal content samples of bighead carp, but only present in one or two samples of grass carp and crucian carp at a very low level (average abundance, <0.02 %). Spirochaetes were absent from all crucian carp samples, but they were present in two grass carp samples (0.39 ± 0.60 %) and one bighead carp sample at 0.10 %. Rare phyla, namely Chlamydiae, TM7, Chlorobi and Deinococcus-Thermus occurred sporadically at low abundance (approximately 0.02 % of the sequences) in some of the samples. The relative abundance of major bacterial phyla in the intestinal content was significantly different between the three species of fish (p < 0.05; Kruskal–Wallis, except for Proteobacteria), Bacteroidetes and Firmicutes were the phyla that exhibited the most significant differences in abundance (p < 0.01; Kruskal–Wallis); in bighead carp, there were more Firmicutes and Proteobacteria than in the two other species. Bacteroidetes were particularly abundant in the gut microbiota of grass carp, and crucian carp was richer in Fusobacteria than the two other carp species.
In the mucosal samples, similarly to what was observed in the intestinal contents, the reads of Fusobacteria, Proteobacteria, Bacteroidetes and Firmicutes were the most abundant ones, accounting in total for 92.54, 99.77 and 98.25 % of the reads in the CB, JB and YB libraries, respectively. Fusobacteria was also the most dominant phylum in the mucosal community. A few other phyla were identified in all mucosal samples, including Spirochaetes (CB, 7.11 %; JB, 0.02 %; and YB, 0.24 %) and Deinococcus-Thermus (CB, 0.14 %; JB, 0.14 %; and YB, 0.06 %).
At the genus level, the sequences from the 13 samples represented 328 genera, with 168 genera found in the water sample, 196 genera in the intestinal content samples (66 genera in grass carp, 54 genera in crucian carp and 152 genera in bighead carp) and 106 different genera in the mucosal samples (37genera in grass carp, 42 genera in crucian carp and 75 genera in bighead carp). The proportion of sequences that could not be assigned to any known genera varied between 0.50 and 32.74 % among the different samples. These sequences, representing 12.63 % of the full dataset, were placed into a total of 114 taxa above the genus level. The representation of unclassified bacterial phylotypes (designated as novel) varied significantly among the subjects (Fig. S3).
The top 10 genera in the water sample were Flavobacterium (46.91 %), Fluviicola (4.37 %), unclassified Comamonadaceae (3.86 %), Albidiferax (3.22 %), unclassified Sphingobacteriales (2.59 %), Arenimonas (2.28 %), Cetobacterium (2.19 %), unclassified Bacteroidetes (1.70 %), unclassified Bacteria (1.56 %) and Undibacterium (1.46 %) by descending order of read abundance.
A total of 28 genera and 22 other taxa (average abundance, >0.1 %; Fig. 2) constituted more than 96 % of the total sequences present in the intestinal content samples. The most abundant genera in grass carp belonged to Cetobacterium (64.51 ± 12.90 %), Bacteroides (14.52 ± 4.85 %), followed by Aeromonas (4.94 ± 3.69 %) and Clostridium XlVa and XlVb (2.06 ± 1.95 and 0.79 ± 0.63 %, respectively). For crucian carp, the community was overwhelmed by Cetobacterium (97.29 ± 0.46 %), while Aeromonas (1.16 ± 0.81 %), Clostridium sensu stricto (0.06 ± 0.11 %), Bacteroides (0.04 ± 0.06 %) and Desulfocapsa (0.01 ± 0.02 %), accounted for low proportions. For bighead carp, the most abundant genera were Cetobacterium (41.16 ± 14.21 %), Clostridium XI (4.56 ± 1.76 %), Clostridium sensu stricto (3.83 ± 2.46 %), Proteocatella (2.81 % ± 0.90 %) and Holdemania (2.39 ± 0.86 %). Our study showed that the genus Cetobacterium constituted the major proportion of microbiota in the intestinal content. In addition, the abundance of the genera mentioned in Fig. 2 was significantly different among the three species (p < 0.05; Kruskal–Wallis, except for Aeromonas, Brevinema, Clostridium sensu stricto, unclassified Betaproteobacteria, unclassified Clostridiales and unclassified Firmicutes). These results indicated that the composition of the bacterial community and the relative abundance of most of the genera might be associated with different physiochemical characteristics in the intestine of the three carp species.

Relative abundance of the predominant genera in the intestinal contents of bighead carp, grass carp and crucian carp
The ten most abundant genera in the mucosal samples were Cetobacterium, Aeromonas, Paludibacter, Brevinema, unclassified Firmicutes, unclassified Bacteroidales, unclassified Vibrionaceae, Clostridium sensu stricto, Grimontia and unclassified Gammaproteobacteria (Table S2), which accounted together for an average of 90.55 %. The members of the genus Cetobacterium were the most numerous in the mucosal communities (CB, 65.81 %; JB, 89.98 %; and YB, 52.46 %).The abundance of Aeromonas, which may include potential pathogens, was significantly different between intestinal content and mucosa (2.13 vs. 6.77 %, respectively; p = 0.05 by a one-tailed Mann–Whitney test).
Core Intestinal Microbiota for Each Species
A major research interest about fish gut communities is to determine whether core microbiota could be delineated for each species, as previously found in other studies [5, 34]. To identify core microbial candidates present in carp intestines, Venn diagrams were constructed. The number of OTUs shared among the intestinal content of the three different individuals was 73, 43 and 239 in grass carp, crucian carp and bighead carp, respectively (Fig. 3a–c). This specific core microbiota represented 10.83, 12.29 and 11.02 % of total OTUs in each of the three species, respectively. Statistical analysis revealed that the OTUs common to the three individuals of each species corresponded to 84.91, 67.06 and 72.53 % of the reads in three grass carp libraries C1, C2 and C3; 86.56, 85.45 and 94.74 % of the reads in three crucian carp libraries J1, J2 and J3; and 58.97, 67.00 and 67.32 % of the reads in three bighead carp libraries Y1, Y2 and Y3, respectively.

Distribution of bacterial genera among the intestinal contents of the individuals of the three fish species. The Venn diagrams (a–c) show the numbers of OTUs (97 % sequence identity) that were shared or unshared by the individuals of grass carp (C1–C3), crucian carp (J1–J3) and bighead carp (Y1–Y3), respectively, depending of overlaps. The pie diagrams (d–f) show the bacterial composition of the intestinal contents of grass carp, crucian carp and bighead carp, respectively. The relative abundances of the bacterial genera present are shown in the chart legend; the figures in parentheses after the legend labels denote the number of OTUs corresponding to the category. The reads that were not identified at the genus level are referred as ‘unclassified’, and the genera that occurred at low abundance were included as ‘others’. The percentages below the pie charts show the contribution of sequences in shared OTUs to all sequences in each species
The three samples from grass carpshared 73 OTUs, out of which 55 belonged to Bacteroides, Clostridium XlVa and XlVb, Cetobacterium, and Aeromonas and accounted together for an average of 70.99 % of total reads (Fig. 3d). For crucian carp, Cetobacterium and Aeromonas included 11.43 % OTUs (40) and 88.65 % reads (16,536) common to the three samples (Fig. 3e). For bighead carp, Acidobacteria Gp17 and Gp6, Paludibacter (Bacteroidetes), Caldilinea (Chloroflexi), Clostridium sensu stricto, Clostridium XI, Holdemania and Proteocatella (Firmicutes), Cetobacterium (Fusobacteria), Blastopirellula, Rhodopirellula and Singulisphaera (Planctomycetes), Dechloromonas (Betaproteobacteria) and Steroidobacter (Gammaproteobacteria) corresponded to 5.03 % of the OTUs and 50.40 % of total reads that were shared by the three individual samples (Fig. 3f).
Relationships Between Fish Gut Communities and Fish-Associated Environmental Bacterial Communities
The PCoA with the weighted UniFrac distance and the heatmap analysis were performed to compare the overall composition of the microbial communities associated with the intestinal contents of the three carps and the environment. The PCoA score plot showed that the water community was remote from the other ones with PC1, which accounted for 49.47 % of the total variation (Fig. 4a). The bacterial communities associated with mucus differed also between species with PC1 and PC3 (Fig. 4a, b). The intestinal mucosal profile was closely related to the highly convergent community from the intestinal contents in crucian carp. The two intestinal content communities of bighead and grass carps were distant from each other along PC2 and PC3, and they were distinct from the respective intestinal mucosal communities (Fig. 4b). Overall, the PCoA plots visualisation explained over 91 % of the variation of microbiotas. In addition, the UniFrac p test showed the highly significant difference between the microbial profiles in the intestinal contents of the three species (Table S3).

Principal coordinates analysis (PCoA) of the dissimilarity between the microbial samples: Weighted UniFrac PCoA plotted against the PC1 vs. PC2 axes (a) and the PC2 vs. PC3 axes (b). The percentages indicate the relative contribution of the three principal components (PC1–PC3)
The hierarchically clustered heatmap analysis based on the bacterial community profiles at the family level disclosed that the bacterial communities in the intestinal content of each species clustered firstly together, and then they clustered with the respective mucosal communities. The composition of intestine microbiota of grass carp and crucian carp shared higher similarity when compared with bighead carp. The water bacterial profile was distant from every intestinal profile (Fig. 5).

Bacterial distribution among the 13 samples. Heatmap based on the hierarchical clustering solution (Bray–Curtis distance metric and complete clustering method) of the 13 samples. Rows represents the 107 predominant bacterial family (average abundance >0.01 %), columns represent the 13 samples, and the values in the heatmap represent the square-root-transformed relative percentage of each bacterial family. The square-root-transformed values for bacterial family are depicted by colour intensity with the legend indicated at the upper right corner of the figure
The bacterial species that were shared by the different communities were determined with a Venn diagram. The number of OTUs shared between the intestinal contents and the mucosal communities were 160, 132, and 238 in the grass carp, crucian carp and bighead carp, respectively (Fig. S4). In other words, 57.14, 52.80 and 58.05 % of the OTUs in the intestinal mucosa library were present in the intestinal content libraries of grass carp, crucian carp and bighead carp, respectively. The most abundant OTUs shared by the two niches in grass carp were Cetobacterium, Aeromonas, Bacteroides and Brevinema (Table S4). Similarly, the most abundant OTUs shared by the two compartments in crucian carp were Cetobacterium, Aeromonas and Clostridium sensu stricto (Table S5). For the bighead carp, the most abundant OTUs shared by the two compartments were Cetobacterium, Clostridium sensu stricto, Paludibactera, Clostridium XI, Dechloromonas and Steroidobacter (Table S6).
Minorities of 48, 41 and 123 OTUs that were present in the water were retrieved in the intestinal contents of grass carp, crucian carp and bighead carp, respectively (Fig. S4). The main genera that were represented both in grass carp and water were Cetobacterium, Aeromonas, Bacteroides and Flavobacterium (Table S7). The most abundant OTUs shared by crucian carp and water communities corresponded to Cetobacterium, Aeromonas and Dechloromonas (Table S8). Concerning bighead carp, the most abundant OTUs also present in water corresponded to Cetobacterium, Dechloromonas, Arenimonas, Gp6 and Steroidobacter (Table S9).
Discussion
Grass carp (C. idellus), crucian carp (C. cuvieri) and bighead carp (H. nobilis) belong to the family of Cyprinidae, and they are currently the three most important species for aquaculture in China. Recent studies have revealed important contributions of gut microbiota to health and disease in vertebrates [35]. The study of the relationships between the composition and diversity of intestinal microbiota and environmental bacterial communities is essential for understanding how gut microbial communities are assembled and how they impact host fitness. Thus far, studies regarding fish intestinal bacterial communities have been relatively limited. To our knowledge, the present study was the first one that addressed the relationships between fish gut bacterial communities of three economically significant species in Chinese aquaculture in the same environment with deep sequencing methodology. Li et al. [14] used DGGE to demonstrate the specificity of bacterial colonisation in several species of Cyprinidae reared in the same hatchery. However, high variability was detected in bacterial profiles associated with individuals from the same species sampled in several rearing laboratory tanks at different times [36]. In the present study, 13 samples from the intestine contents and mucosa of three carps and pond water were analysed by using pyrosequencing of the V4/V5 regions of the 16S rRNA gene.
The PCoA score plot and heatmap analysis revealed clearly that the intestinal bacterial communities of the three species of fish harboured specific features when they are cohabiting in the same environment. The bacterial communities associated with the intestinal mucosa were different from those of the intestinal contents, and the water samples were further dissimilar. These results suggested the presence of specific bacterial populations in the different species. Given that these fish were reared under the same environmental conditions, these results suggest that the gut microbiota composition of fishes is not a simple reflection of the microorganisms in their local habitat, but result from selective pressures within the gut, depending on species-specific behaviour, immunity and metabolism, as recently shown in other species [37, 38].
The gut microbial analysis demonstrated that the dominant bacteria of the three species of fish belonged to four phyla, Fusobacteria, Proteobacteria, Bacteroidetes and Firmicutes, which are commonly encountered in the vertebrate gastrointestinal tract [17]. These results are consistent with earlier study on freshwater Teleosts [5, 12, 14, 39], including grass carp and common carp (Cyprinus carpio). Almost all Fusobacteria detected in the present fish intestinal samples belonged to the genus Cetobacterium (47,728 out of 50,809 reads, i.e. 93.94 %). It has been shown that Cetobacterium can produce vitamin B12 at high efficiency and ferment peptides and carbohydrates [40]. This may give Cetobacterium a particular relevance to fish nutrition. Only a limited number of studies have reported Cetobacterium as the dominant microbiota in fish intestine [39, 41]. PCR biases might be suspected to cause this discrepancy, but the comparison with the RDP databases showed that the primers used here were more sensitive to Firmicutes than to Fusobacteria (Table S10). These results confirmed thus that Cetobacterium are naturally abundant in the gut of carps. Crucian carp had much higher abundance of Cetobacterium (>90 %), compared with the other two species in the present study. Another species of the genus Carassius, gibel carp (Carassius auratus gibelio), had the relative abundance of Cetobacterium increased 7–11-fold in gut microbiota, when the carp was cultivated at high stocking density, a common practice in China to fully utilise the ponds, and to maximise yield [42]. A possible explanation for the overwhelming abundance of Cetobacterium in crucian carp may come from its high stocking density of this fish in the present study (10 to 15 kg/m−3).
Besides Fusobacteria, Bacteroidetes and Firmicutes were the phyla that exhibited the most significant differences in abundance among the three species of fish (p < 0.01; Kruskal–Wallis). The proportion of Bacteroidetes was higher in grass carp than in the two other species, while Firmicutes were particularly abundant in bighead carp. Among Bacteroidetes, members of the genera Bacteroides and Paludibacter were the most abundant in the gut microbiota of grass carp and bighead carp, respectively (including intestinal content and mucosa). Both Bacteroides and Paludibacter may play an important role in fish intestine, due to the fermentation of plant-derived substrates in the gut of carps, allowing them to maximise the energy intake from indigestible components by producing high levels of short-chain fatty acids (SCFA) that supply the host with an additional amount of energy [3, 7, 39, 43, 44]. Among Firmicutes, Clostridium XlVa and XlVb were dominant in the intestinal microbiota of grass carp. In bighead carp, Clostridium sensu stricto (cluster I), Clostridium XI and Proteocatella were the most abundant Firmicutes. Both Clostridium XlVa and XlVb are versatile in their ability to utilise various polysaccharides, such as cellulose, xylan and hemicelluloses, which constitute the major part of vegetalfibres [45, 46]. By contrast, members of the genera Clostridium sensu stricto (cluster I) and Clostridium XI include not only species with saccharolytic and fibre-fermenting activities but also proteolytic species [47, 48]. Proteocatella includes fermentative proteolytic bacteria that are able to use various products of proteolysis as substrates but not sugars [49]. The three carp species differ by their natural feeding habits, and their feed intake was likely different, even though commercial food was supplied in the pond. Grass carp are herbivorous, and crucian carp are omnivorous with a tendency to herbivory. Therefore, plant polysaccharide-degrading bacteria are particularly important for food degradation in the intestine of these carps [12]. Contrastingly, bighead carp are filter-feeders, preferentially consuming zooplankton, but also phytoplankton and detritus. This filter-feeding activity resulted in a much higher bacterial diversity in the intestinal content of bighead carp, compared with those of the two grazing species [50, 51]. The composition of intestinal microbiota of the two grazing species, grass carp and crucian carp, shared higher similarity between them, compared with bighead carp.
Proteobacteria are known members of the gut microbiota of many organisms including fish [6, 14, 39]. Most Proteobacteria found in carps belonged to Aeromonas, which include potential pathogens to fishes [6, 7]. Members of the genus Aeromonas were found mainly in grass carp and crucian carp in this study. This finding is in accordance with a former study that suggested that fish gut may be a reservoir for many opportunistic pathogens [52]. The genus Aeromonas was significantly more abundant in the intestinal mucosa than in the intestinal content. These observations are consistent with previous studies reporting that Aeromonas with adhesive capability can colonise the surface of intestinal mucosa and that the intestine might be a primary location for stress-induced infections by Aeromonas [53]. However, bacteria associated with the mucosa may be regarded as indigenous species, and they are involved in host nutrition, mucosal defence and host immunity [54]. Although several Aeromonas strains are potential pathogens, Aeromonas have been detected in the normal intestinal mucosa from many fishes [5–7, 12]. A high concentration of Aeromonas in the gut of healthy fish may significantly contribute to the digestive function [7].
Interestingly, the RDP Classifier was unable to classify an average of 12 % of sequences to the genus level. The phylotypes in this category are not in the preexisting taxonomic framework of RDP database, suggesting that most of them likely represent novel bacteria. Although a limited number of unclassified sequences can result from PCR errors or sequencing artefacts, such an abundance of unclassified reads argues for a significant presence of novel species. Our discovery of numerous novel bacteria in the intestine is consistent with massive identification of novel species in the intestinal flora of fish [6, 10]. These results suggest that fish gut harbours a larger bacterial diversity than previously recognised. A detailed phylogenetic characterization of unclassified sequences and their phylogeny is an interesting direction of future research.
The concept ‘core microbiota’ referred to a set of abundant microbial lineages that are shared by all individuals from the same species [19]. The concept of core gut microbiota has first been explored in the context of mammalian hosts [17, 34], and then in freshwater fish [5, 12, 19]. The bacterial profiles observed in the three species of carps may thus reflect specific core microbiotas, beyond the differences attributable to feeding behaviour, which was especially visible in the intestinal contents of filter-feeding bighead carp in the present analyses. These data and previous studies indicate that Fusobacteria, Firmicutes, Proteobacteria and Bacteroidetes were the main phyla represented in the specific bacterial cores associated with the intestinal contents of grass carp and bighead carp, while the intestinal core microbiota of crucian carp appeared less diversified, with Fusobacteria and Proteobacteria as the main phyla. We found that the three individuals of each species analysed in this study harboured very similar intestinal bacterial communities dominated by specific core microbiota, which appeared larger shared core OTUs than those described in previous studies (Fig. 3a–c) [5, 17]. All the carps analysed here were obtained from a single commercial supplier and raised under identical husbandry conditions, thereby limiting the environmental variation and increasing the likelihood of similar microbial profiles. Further comparisons of gut microbiota from carps obtained from different aquaculture facilities or caught in the wild will be necessary to delineate the ‘true’ core microbiota shared by carps of the same species.
It may be hypothesised that the ancestral members of intestinal microbiota settled in the gastrointestinal tract and co-evolved with their hosts, notably in response to feeding habits, and to immune tolerance. This interdependence shapes likely the gut bacterial communities in fish. The diet and the environment affect the intestinal microbiota of fish and mammals [2, 8, 13, 47], but the same environment in the present study did not result in similar intestinal bacteria, suggesting that the specific endogenous factors outweighed by far the environmental factors to mould the composition of microbiota. The species-specific selective pressures in fish gut may be related to the genetic features of the host that govern behaviour and metabolism. Such genetic influence has been mainly studied for the impact on Firmicutes and Bacteroidetes, which can ferment carbohydrates and/or proteins fermentation in the intestine, helping thus the host to acquire nutrients from the diet [55–57].
References
Walter J, Britton RA, Roos S (2011) Host-microbial symbiosis in the vertebrate gastrointestinal tract and the Lactobacillus reuteri paradigm. Proc Natl Acad Sci U S A 108:4645–4652
Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6:776–788
Flint HJ, Bayer EA, Rincon MT, Lamed R, White BA (2008) Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol 6:121–131
Sugita H, Miyajima C, Deguchi Y (1991) The vitamin B12-producing ability of the intestinal microflora of freshwater fish. Aquaculture 92:267–276
Roeselers G, Mittge EK, Stephens WZ, Parichy DM, Cavanaugh CM, Guillemin K, Rawls JF (2011) Evidence for a core gut microbiota in the zebrafish. ISME J 5:1595–1608
Wu S, Gao T, Zheng Y, Wang W, Cheng Y, Wang G (2010) Microbial diversity of intestinal contents and mucus in yellow catfish (Pelteobagrus fulvidraco). Aquaculture 303:1–7
Nayak SK (2010) Role of gastrointestinal microbiota in fish. Aquac Res 41:1553–1573
Sullam KE, Essinger SD, Lozupone CA, O’CONNOR MP, Rosen GL, Knight R, Kilham SS, Russell JA (2012) Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol 21:3363–3378
Greetham H, Giffard C, Hutson R, Collins M, Gibson G (2002) Bacteriology of the Labrador dog gut: a cultural and genotypic approach. J Appl Microbiol 93:640–646
Kim DH, Brunt J, Austin B (2007) Microbial diversity of intestinal contents and mucus in rainbow trout (Oncorhynchus mykiss). J Appl Microbiol 102:1654–1664
Spanggaard B, Huber I, Nielsen J, Nielsen T, Appel K, Gram L (2000) The microflora of rainbow trout intestine: a comparison of traditional and molecular identification. Aquaculture 182:1–15
Wu S, Wang G, Angert ER, Wang W, Li W, Zou H (2012) Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS One 7:e30440. doi:10.1371/journal.pone.0030440
Navarrete P, Magne F, Araneda C, Fuentes P, Barros L, Opazo R, Espejo R, Romero J (2012) PCR-TTGE analysis of 16S rRNA from rainbow trout (Oncorhynchus mykiss) gut microbiota reveals host-specific communities of active bacteria. PLoS One 7:e31335. doi:10.1371/journal.pone.0031335
Li X, Yu Y, Feng W, Yan Q, Gong Y (2012) Host species as a strong determinant of the intestinal microbiota of fish larvae. J Microbiol 50:29–37
Cardenas E, Tiedje JM (2008) New tools for discovering and characterizing microbial diversity. Curr Opin Biotechnol 19:544–549
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers allow hundreds of samples to be pyrosequenced in multiplex. Nat Methods 5:235
Nam Y-D, Jung M-J, Roh SW, Kim M-S, Bae J-W (2011) Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS One 6:e22109. doi:10.1371/journal.pone.0022109
Ravussin Y, Koren O, Spor A, LeDuc C, Gutman R, Stombaugh J, Knight R, Ley RE, Leibel RL (2012) Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 20:738–747
Wong S, Waldrop T, Summerfelt S, Davidson J, Barrows F, Kenney PB, Welch T, Wiens GD, Snekvik K, Rawls JF (2013) Aquacultured rainbow trout (Oncorhynchus mykiss) possess a large core intestinal microbiota that is resistant to variation in diet and rearing density. Appl Environ Microbiol 79:4974–4984
MoA BoF (2011) China fisheries statistical yearbook 2011. China Agriculture Press, Beijing
BillaRd R, BeRni P (2004) Trends in cyprinid polyculture. Cybium 28:255–261
Ringø E, Sperstad S, Myklebust R, Refstie S, Krogdahl Å (2006) Characterisation of the microbiota associated with intestine of Atlantic cod (Gadus morhua L.): the effect of fish meal, standard soybean meal and a bioprocessed soybean meal. Aquaculture 261:829–841
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522
Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35:e120
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Schloss PD (2009) A high-throughput DNA sequence aligner for microbial ecology studies. PLoS One 4:e8230
Gaspar JM, Thomas WK (2013) Assessing the consequences of denoising marker-based metagenomic data. PLoS One 8:e60458. doi:10.1371/journal.pone.0060458
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Claesson MJ, O'Sullivan O, Wang Q, Nikkilä J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O'Toole PW (2009) Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4:e6669. doi:10.1371/journal.pone.0006669
Lozupone C, Hamady M, Knight R (2006) UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinforma 7:371. doi:10.1186/1471-2105-7-371
Kemp PF, Aller JY (2004) Estimating prokaryotic diversity: when are 16S rDNA libraries large enough? Limnol Oceanogr Methods 2:114–125
Stackebrandt E, Goebel B (1994) Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44:846–849
Piao Z, Yang L, Zhao L, Yin S (2008) Actinobacterial community structure in soils receiving long-term organic and inorganic amendments. Appl Environ Microbiol 74:526–530
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484
Sekirov I, Russell SL, Antunes LCM, Finlay BB (2010) Gut microbiota in health and disease. Physiol Rev 90:859–904
Gatesoupe FJ, Covès D, Ortega A, Papandroulakis N, Vadstein O, Gándara F (2013) A spatiotemporal study of bacterial community profiles associated with Atlantic bluefin tuna larvae, Thunnus thynnus L., in three Mediterranean hatcheries. Aquac Res 44:1511–1523
Larsen AM, Mohammed HH, Arias CR (2014) Characterization of the gut microbiota of three commercially valuable warmwater fish species. J Appl Microbiol 116:1396–1404
Ye L, Amberg J, Chapman D, Gaikowski M, Liu W-T (2013) Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J 8:541–551
van Kessel MA, Dutilh BE, Neveling K, Kwint MP, Veltman JA, Flik G, Jetten MS, Klaren PH, den Camp HJO (2011) Pyrosequencing of 16S rRNA gene amplicons to study the microbiota in the gastrointestinal tract of carp (Cyprinus carpio L.). AMB Express 1:1–9. doi:10.1186/2191-0855-1-41
Finegold SM, Vaisanen M-L, Molitoris DR, Tomzynski TJ, Song Y, Liu C, Collins MD, Lawson PA (2003) Cetobacterium somerae sp. nov. from human feces and emended description of the genus Cetobacterium. Syst Appl Microbiol 26:177–181
Ni J, Yu Y, Zhang T, Gao L (2012) Comparison of intestinal bacterial communities in grass carp, Ctenopharyngodon idellus, from two different habitats. Chin J Oceanol Limnol 30:757–765
Zhou, Z., He, S., Liu, Y., Shi, P., Yao, B., Ringø, E. (2012). Do stocking densities affect the gut microbiota of gibel carp Carassius auratus gibelio cultured in ponds? J Aquacult Res Development 3. doi:10.4172/2155-9546.S1-003
Dominika Ś, Arjan N, Karyn RP, Henryk K (2011) The study on the impact of glycated pea proteins on human intestinal bacteria. Int J Food Microbiol 145:267–272
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107:14691–14696
Uffen R (1997) Xylan degradation: a glimpse at microbial diversity. J Ind Microbiol Biotechnol 19:1–6
Uz I, Ogram AV (2006) Cellulolytic and fermentative guilds in eutrophic soils of the Florida Everglades. FEMS Microbiol Ecol 57:396–408
Schwab C, Cristescu B, Northrup JM, Stenhouse GB, Gänzle M (2011) Diet and environment shape fecal bacterial microbiota composition and enteric pathogen load of grizzly bears. PLoS One 6:e27905. doi:10.1371/journal.pone.0027905
Lubbs D, Vester B, Fastinger N, Swanson K (2009) Dietary protein concentration affects intestinal microbiota of adult cats: a study using DGGE and qPCR to evaluate differences in microbial populations in the feline gastrointestinal tract. J Anim Physiol Anim Nutr 93:113–121
Pikuta EV, Hoover RB, Marsic D, Whitman WB, Lupa B, Tang J, Krader P (2009) Proteocatella sphenisci gen. nov., sp. nov., a psychrotolerant, spore-forming anaerobe isolated from penguin guano. Int J Syst Evol Microbiol 59:2302–2307
Verschuere L, Rombaut G, Sorgeloos P, Verstraete W (2000) Probiotic bacteria as biological control agents in aquaculture. Microbiol Mol Biol Rev 64:655–671
Tetlock A, Yost CK, Stavrinides J, Manzon RG (2012) Changes in the gut microbiome of the sea lamprey during metamorphosis. Appl Environ Microbiol 78:7638–7644
Pond MJ, Stone DM, Alderman DJ (2006) Comparison of conventional and molecular techniques to investigate the intestinal microflora of rainbow trout (Oncorhynchus mykiss). Aquaculture 261:194–203
Namba A, Mano N, Hirose H (2007) Phylogenetic analysis of intestinal bacteria and their adhesive capability in relation to the intestinal mucus of carp. J Appl Microbiol 102:1307–1317
Ringø E, Olsen RE, Mayhew TM, Myklebust R (2003) Electron microscopy of the intestinal microflora of fish. Aquaculture 227:395–415
Spor A, Koren O, Ley R (2011) Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9:279–290
Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS (2009) Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A 106:15813–15818
Palma G, Capilla A, Nadal I, Nova E, Pozo T, Varea V, Polanco I, Castillejo G, López A, Garrote JA (2010) Interplay between human leukocyte antigen genes and the microbial colonization process of the newborn intestine. Curr Issues Mol Biol 12:1–10
Acknowledgements
This work was supported by grants from the National Science and Technology Pillar Programmes (grants no. 2011BAI15B01-41), FEBL project (2011FBZ26) and the Natural Science Foundation of China (no. 30670112 and 31070112).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 346 kb)
Rights and permissions
About this article

Cite this article
Li, T., Long, M., Gatesoupe, FJ. et al. Comparative Analysis of the Intestinal Bacterial Communities in Different Species of Carp by Pyrosequencing. Microb Ecol 69, 25–36 (2015). https://doi.org/10.1007/s00248-014-0480-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-014-0480-8