
Abstract
Ectomycorrhizal fungi are surrounded by bacterial communities with which they interact physically and metabolically during their life cycle. These bacteria can have positive or negative effects on the formation and the functioning of ectomycorrhizae. However, relatively little is known about the mechanisms by which ectomycorrhizal fungi and associated bacteria interact. To understand how ectomycorrhizal fungi perceive their biotic environment and the mechanisms supporting interactions between ectomycorrhizal fungi and soil bacteria, we analysed the pairwise transcriptomic responses of the ectomycorrhizal fungus Laccaria bicolor (Basidiomycota: Agaricales) when confronted with beneficial, neutral or detrimental soil bacteria. Comparative analyses of the three transcriptomes indicated that the fungus reacted differently to each bacterial strain. Similarly, each bacterial strain produced a specific and distinct response to the presence of the fungus. Despite these differences in responses observed at the gene level, we found common classes of genes linked to cell–cell interaction, stress response and metabolic processes to be involved in the interaction of the four microorganisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhizospheres form a complex ecosystem in which roots interact with communities of highly diverse microorganisms, which is known as the microbiome [1, 2]. Similar to the human gut microbiome which can impact fundamental host processes ranging from physiology to immunology [3], plants cope with microorganisms living inside and outside their tissues and rely on some for nutrient acquisition and defence against pests [4]. Ectomycorrhizal fungi play a particularly important role by providing mineral nutrients in exchange for carbohydrates and by stimulating the plant’s defence systems [5]. As a part of the rhizosphere microbiome, ectomycorrhizal fungi themselves are permanently surrounded by numerous microorganisms including bacteria with which they interact both physically and metabolically [6]. Our knowledge is relatively limited about how ectomycorrhizal and bacterial consortia establish and maintain themselves as well as the identity and characteristics of molecular interactions between these microorganisms. Ectomycorrhizal fungi perceive the presence of host roots and are chemoattracted towards these roots [7]. Ectomycorrhizal fungi also modulate their physiology and respond at the transcription level to the presence of mycorrhiza helper bacteria which stimulate mycorrhiza formation [8–11]. However, whether the fungal response is specific to beneficial bacteria or extends towards neutral or detrimental bacteria is not known. Surprisingly, relatively little is known about the molecular mechanisms involved in the interaction between ectomycorrhizal fungi and bacteria. To understand the specificities of these molecular pathways, we conducted a simultaneous analysis of transcriptomic responses of the ectomycorrhizal fungus Laccaria bicolor S238 with three soil bacteria during their interactions. The three different soil bacterial strains were chosen for their contrasting effects on the growth and morphology of the fungus [8]: Pseudomonas fluorescens BBc6R8, a mycorrhiza helper strain of L. bicolor S238N which had been isolated from a sporocarp of the fungus [8], P. fluorescens Pf29Arp, a strain commensal to L. bicolor S238N [12, 13] and Collimonas fungivoransTer331, a strain antagonistic to L. bicolor S238N [8, 14]. While strain BBc6R8 stimulates radial growth, the apex density and the branching angle and density of the fungal mycelium, strain Pf29Arp only enhances the branching angle of the hyphae. In contrast, the antagonist strain Ter331 inhibits radial growth of the fungus but stimulates hyphal branching and density. In parallel, we also analysed the differential responses of the three bacterial strains to the presence of the ectomycorrhizal fungus at the transcriptomic level to test whether and how the bacteria reacted to the interaction with the fungus.
Materials and Methods
Fungal–Bacterial Confrontation Bioassay
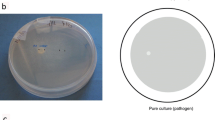
The ectomycorrhizal basidiomycete L. bicolor S238N was maintained on Pachlewski agar medium P5 [15] at 25 °C for 3 weeks. P. fluorescens BBc6R8, P. fluorescens Pf29Arp and C. fungivorans were stored at −80 °C in LB medium [16] with 20 % glycerol added. To prepare the bacterial inocula for the in vitro bioassays with L. bicolor, the bacterial strains were first grown on 10 % TSA plates (tryptic soy broth from Difco and 15 g l−1 of agar) at 25 °C for 65 h. The bioassay was prepared as described by Deveau et al. [8]. A plug of L. bicolor S238N was cut out from the edge of a colony grown on P5 medium (0.5 g Di-NH4 + tartrate, 1 g KH2PO4, 0.5 g MgSO4, 20 g glucose, 5 g maltose, 1 ml 1/10 diluted Kanieltra microelement solution, 1 ml thiamine 100 mg/l−1 solution, and 20 g agar l−1 at pH 5.5 for 1 l) and transferred into the centre of a P20Th– plate (0.5 g Di-NH4 + tartrate, 1 g KH2PO4, 0.5 g MgSO4, 1 g glucose, 1 ml 1/10 diluted Kanieltra microelement solution, and 20 g agar l−1 at pH 5.5 for 1 L). Four 10-μl droplets bacterial suspension (OD600 nm 0.7 in sterile water) were distributed at 1.2 cm from the centre of the fungal plug of mycelium (Fig. 1). Control treatments were performed, either without fungus or where bacteria were replaced by sterile water droplets. Plates were sealed with plastic tape and incubated at 10 °C in the dark. For each fungal treatment and corresponding mock control, mycelia of L. bicolor S238N from 50 plates were collected, frozen in liquid nitrogen and pooled. For each bacterial treatment and corresponding mock control, half of the four colonies facing the mycelium were collected from ten plates, frozen in liquid nitrogen and pooled. Fungal and bacterial samples were collected from independent plates to avoid changes in transcriptomic responses caused by handling of the plates during the isolation of the interacting partner. The full experiment was performed three times independently.

Scheme of the in vitro confrontation assay and of the sampling strategy for transcriptomic analyses. Four droplets of bacterial suspension (white circles) were distributed at 1.2 cm from the center of the fungal plug of mycelium (grey circle). Control treatments without fungus (control 1) or in which bacteria were replaced by sterile water droplets (control 2) were performed. For each fungal treatment and corresponding mock control, mycelium of L. bicolor S238N from 50 plates was collected, frozen in liquid nitrogen and pooled. For each bacterial treatment and corresponding mock control, the half part of the four colonies facing the mycelium was collected from ten plates, frozen in liquid nitrogen and pooled
Fungal RNA Isolation and cDNA L. bicolor S238N Array Hybridization
After 14 days of incubation in the dark at 10 °C, the whole mycelium from 50 plates was collected, frozen in liquid nitrogen, pooled and stored at −80 °C. RNA from 50 mg of frozen mycelium was then extracted using the RNeasy Plant mini kit (Qiagen, Hilden, Germany) as recommended by the manufacturer for fungal material. Quality of RNA was checked by RNAse-free 1 % agarose electrophoresis. Synthesis of complex cDNA probes from 400 ng of fungal RNA and cDNA hybridization on a Nylon membrane containing 4,992 cDNAs were performed as described in Deveau et al. [8]. Identification of cDNA clones corresponding to up- and down-regulated transcripts was carried out as described by Deveau et al. [8].
Validation of Expression Array Data
Validity and reproducibility of array data were checked by calculating the Pearson correlation coefficient which was over 0.95 for all set of arrays (data not shown). Data quality assessment was performed through analysis of variance (t-test), and a Bayesian statistical framework was implemented in the Cyber-T web interface [17] as described by Duplessis et al. [18]. Regulated transcripts were identified by the following criteria: (1) when significant modification (Bayesian t-test) of gene expression was detected in at least two of the three replicates and (2) when the average of fold changes (treatment vs. control) in the expression level was observed at plus or minus twofolds (Duplessis et al., 2005). A Posterior Probability of Differential Expression test (PPDE test) was used to determine the global false positive and negative levels. The complete expression data set is available as a series (accession numbers GSE53608 and GSE53609) at the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/). Finally, quantitative RT-PCR measurements were performed on five fungal transcripts with altered levels of expression in response to at least one of the three bacterial strains (tectonin 2, hypothetical protein 456319, hypothetical protein 656382, glutathione-S-transferase 674829, hypothetical protein 304792). Eight non-regulated genes were tested as controls and the most stable of the two (hypothetical protein 701050-CloneLB17E10 and trehalose phosphorylase) were used for data normalization as previously described by Deveau et al. [8]. Primer design and qRT-PCR were performed as described by Deveau et al. [8]. Primer sequences are provided in Table 1.
Bacterial RNA Isolation
All samples were collected after 14 days of incubation before contact occurred between bacterial cells and fungal hyphae. The semicircular section of the bacterial colonies from P. fluorescens BBc6R8, Pf29Arp or C. fungivorans Ter331 which faced the mycelium (Fig. 1) was collected from ten plates, frozen in liquid nitrogen and then pooled. Three independent biological replicates per treatment were performed except for C. fungivorans Ter331 for which two replicates were obtained. RNA was then extracted using Trizol (Invitrogen, Stockholm, Sweden) as recommended by the manufacturer. One hundred micrograms of total RNA were further purified using the RNeasy Plant mini kit (Qiagen, Hilden, Germany) as recommended by the manufacturer. Quality of RNAs was checked by electrophoresis using an Experion automated electrophoresis system (Biorad, USA). RNAs were then used for hybridization against a microarray specific for BBc6R8 (this paper), Ter331 [19] or Pf29Arp [20].
BBc6R8 Microarray Design, Hybridization and Analysis
BBc6R8 cDNA microarray synthesis, array hybridization and scanning were performed by NimbleGen (Roche NimbleGen Systems, Iceland). The cDNA were synthesized from 20 μg total RNA. The P. fluorescens BBc6R8 NimbleGen expression array was designed based on the initial genome annotation (available on Genbank—referenced under accession numbers AKXH01000001–AKXH01000163). The array was designed as follows: four oligomers of 60-nucleotides-long probes were designed for 6,469 of the predicted open reading frames. For each probe, three replicates were synthesized on the array. Probes for 16 genes from L. bicolor S238N were then added and used to quantify the background signal. BBc6R8 cDNA microarray synthesis, array hybridization and scanning were performed by NimbleGen (Roche NimbleGen Systems, Iceland). The cDNA were synthesized from 20 μg total RNA. Average expression levels were calculated for each gene from the independent probes on the array and then used for further analysis. Raw array data were normalized by the robust multiarray average (RMA) routine using the ARRAYSTAR software (Dnastar Inc., Madison, WI, USA). A Student’s t-test with FDR correction (Benjamini–Hochberg) was applied to the data using ARRAYSTAR. Transcripts with a significant P-value (<0.05) and ≥ twofold change in transcript level were considered to be differentially expressed. The complete expression data set is available as a series (accession number GSE38243) on the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/).
Pf29A DNA Microarray Hybridization and Data Analysis
Shotgun DNA microarrays used are fully described in Barret et al. [20]. Hybridization, scan and analyses were performed as previously described [20]. The contribution of each experimental factor (gene, fungus, biological replicate, dye) and the interactions between them were evaluated using a global ANOVA. Differentially expressed genes were selected by imposing stringent cutoffs (see “Results” section) on the local ANOVA graphic of the interaction ‘gene–fungus’.
The complete expression data set is available as a series (accession number GSE55147) on the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/).
Ter331 Microarray Hybridization and Data Analysis
Ter331 cDNA microarray synthesis, array hybridization and scanning were performed by NimbleGen (Roche NimbleGen Systems, Iceland) as previously described in Mela et al. [19]. The cDNA were synthesized from 20 μg total RNA. Chip information is available via the MIAMExpress database (accession number A-MEXP-1876). Average expression levels and raw data analysis were the same as for BBc6R8 analysis. The complete expression data set is available as a series (accession number GSE54533) on the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/).
Results
Bacterial Strains Differentially Impact Gene Expression in L. bicolor S238N
In S238N, 22, 31 and 83 cDNAs representing 16, 14 and 70 transcripts were found to be differentially expressed (≤or > twofold) in the presence of P. fluorescens BBc6R8, Pf29Arp and C. fungivorans Ter331, respectively, compared to unconfronted cultures of the fungus (Table 2). Levels of regulation varied between two- and 11folds, the highest levels of regulation being observed for strain C. fungivorans Ter331. All S238N transcripts were up-regulated by P. fluorescens BBc6R8 and Pf29Arp, while 12 and 58 transcripts were up- and down-regulated in response to C. fungivorans Ter331, respectively. Functional categories of S238N transcripts regulated by the three bacteria are presented in Fig. 2. A Venn diagram was used to visualize how similar the responses of L. bicolor S238N were between the three types of interactions (Fig. 3). Most of the transcripts fell into the outer “unique” zones of the Venn diagram, implying that their regulations were strain specific. Only three transcripts were co-regulated by the different bacterial strains. Those three responsive transcripts encoded for a metallothionein (transcript ID 388066), a small hypothetical protein of 51 amino acids specific to L. bicolor S238N (308883) and a hypothetical protein Tra1 (456319) potentially involved in transcription regulation (Table 2). All were down-regulated in response to Ter331 and up-regulated in response to BBc6R8 and Pf29Arp. A transcript encoding for tectonin 2 was up-regulated in the presence of BBc6R8 and Pf29Arp. The transcript was not found to be significantly up-regulated in the presence of Ter331 on arrays because of high variability in the signal intensity between arrays for this transcript. However, quantitative RT-PCR indicated that it was also overexpressed in response to Ter331 (t-test, P < 0.01; Table 2). In addition, two transcripts encoding two hypothetical proteins only found so far in the genome of L. bicolor S238N (608752 and 656382) were co-regulated in the presence of BBc6R8 and Pf29Arp (Fig. 3). Transcripts induced only by BBc6R8 were involved in transcription (splicing factor 3b), chromatin structure (histone H4), stress response (one other metallothionein) and transport (major facilitator super family 1 transport protein), while transcripts responsive to Pf29Arp were predicted to encode proteins whose function was unknown (Table 2). Finally, Ter331 induced the down-regulation of several transcripts involved in protein synthesis (e.g. ribosomal proteins, translation elongation factor), mitochondrial activity (e.g. cytochrome c oxidases, ADP/ATP carrier, coenzyme Q biosynthesis) and stress response (glutathione-S-transferases). Two transcripts encoding proteins potentially involved in the regulation of cell wall integrity (pil1) and response to pH (predicted protein with PalI/Rim9 domain) were down-regulated. Three other transcripts encoding proteins potentially involved in signal transduction were either up- (phosphoesterase) or down-regulated (protein kinases). Validation of regulation levels of five S238N-responsive transcripts was performed by quantitative RT-PCR (Fig. 4).

Gene ontology of L. bicolor S238N transcripts differentially expressed in the presence of the three bacterial strains. Data are expressed as the number of transcripts down-regulated (open) or up-regulated (closed) in the presence of strains P. fluorescens BBc6R8, Pf29Arp, C. fungivorans Ter331

Venn diagram showing the number of transcripts differentially expressed in L. bicolor S238N (a) in the presence of P. fluorescensBBc6R8, Pf29Arp, C. fungivorans Ter331 and in the three bacterial strains (b) in the presence of L. bicolor S238N. Values represent the number of transcripts that were >2-fold induced or repressed during the interaction compared to control treatment without interacting microorganisms. Intersects show the number of transcripts which were regulated in response to several bacterial strains

Quantitative RT-PCR validation of macroarray data. Comparison of the expression ratio [bacterial treatment/water control] measured by quantitative PCR (black) or macroarray analyses (grey). Each value represents the average value of three biological replicates ± standard error
Bacterial Strains React Differently in the Presence of L. bicolor S238N at the Transcriptomic Level
Twenty-nine and 14 transcripts from strain BBc6R8 and Ter331, and seven genomic regions corresponding to 12 transcripts of Pf29Arp were found to be significantly regulated after 14 days of incubation with L. bicolor S238N, respectively. Regulation levels were observed as no higher than 12 folds. All genes differentially regulated from strain Pf29Arp in the presence of L. bicolor S238N were down-regulated, while expression of 16 and 13 genes was down-regulated in strains BBc6R8 and Ter331, respectively. Moreover, the expression of four and ten genes of BBc6R8 and Ter331, respectively, were up-regulated in the presence of S238N. Regulated transcripts in the three strains were distributed in seven functional categories (Fig. 5, Table 3): primary metabolism (nine transcripts), cell envelop biogenesis (eight), transport (seven), transcription regulation (seven), DNA replication and repair (two), other functions (seven) and hypothetical proteins (13). No orthologous gene was commonly regulated between strains. However, some functional categories were shared between bacteria: transcription factors were regulated in all strains but none was encoding for the same type of transcription regulator (Table 3). Regulation of the transcription of genes involved in primary metabolism was observed in all strains. Transcripts involved in lipid metabolism (cyclopropane-fatty-acyl-phospholipid synthase), arginine catabolism (carbamate kinase) and NAD production (nicotinatephosphoribosyltransferase, NAD synthetase) were up-regulated in BBc6R8, while transcript involved in amino acid metabolism were down-regulated in Pf29Arp (B12-dependent methionine synthase) and Ter331 (saccharopine dehydrogenase). In addition, transcripts encoding proteins related to transport of nutrients were regulated in BBc6R8 and Pf29Arp: transcripts encoding for one MFS transporter, an oligopeptide transporter (Pf29Arp) and a glucose ABC transporter (BBc6R8) were down-regulated, while one transcript encoding a glycine/betain ABC transporter was up-regulated in BBc6R8. Finally, several transcripts encoding proteins which could be associated to cell membrane biogenesis were regulated in the three strains: observed among the most highly up-regulated transcripts (seven fold) of BBc6R8 was a cluster of four genes (PfR8_560-565) related to capsular polysaccharide biosynthesis, while one transcript associated with membrane-derived oligosaccharide synthesis was down-regulated in Pf29Arp as well as an endochitinase potentially involved in cell wall remodelling in Ter331.

Gene ontology of P. fluorescens BBc6R8, Pf29Arp, C. fungivorans Ter331 transcripts differentially expressed in the presence of L. bicolor S238N. Data are expressed as the number of transcripts down-regulated (open) or up-regulated (closed) in the presence of L. bicolor S238N
Discussion
Ectomycorrhizal fungi interact with bacterial strains which can have beneficial, neutral or harmful effects on fungal physiology. Mechanisms by which ectomycorrhizal perceive and react to surrounding microorganisms are to date underexplored. However, previous related studies have brought out attention to a molecular dialog involving recognition processes within the ectomycorrhizal biotic environment by the fungus and subsequent production of an adapted response [8, 9, 11]. To determine how this molecular dialog occurs between the microorganisms and its degree of specificity, we performed a dual-transcriptomic analysis of the interaction between the ectomycorrhizal fungus L. bicolor S238N and three soil bacterial strains related to the Pseudomonas fluorescens and Collimonas genus. Bacteria belonging to P. fluorescens group were enriched in the mycorrhizosphere of L. bicolor [21], while Collimonas strains were detected in forest soils typically colonized by L. bicolor [14, 22]. These bacterial strains were chosen for their contrasting effects on the growth and morphology of L. bicolorS238N [8]. This experiment allowed us to further characterize transcriptomes from the four microorganisms while comparing their behavioural patterns. To the best of our knowledge, this is the first study in which the transcriptomes from both an ectomycorrhizal fungus and interacting bacteria have been analysed at the same time.
Although changes in L. bicolor transcriptome were observed in response to the presence of the three bacterial strains, the strain Ter331 modified the expression of numerous fungal genes when compared to BBc6R8 and Pf29Arp strains. Contrasting responses were observed, suggesting that the ectomycorrhizal fungus responded specifically to each bacterium. Such differential response had previously been described in the case of the interaction between filamentous bacterial Streptomyces strains [23]. Highly specific interactions between Aspergillus nidulans and Streptomyces bacteria were also observed [24], reinforcing the hypothesis that microbes have the potential to precisely recognize their biotic environment and are able to react to it accordingly. Our data suggests that the ectomycorrhizal fungus L. bicolor S238N might also have this ability. Interestingly, even two closely related strains belonging to the same taxonomic group induced different morphological and transcriptomic responses, suggesting that this process is strain specific. Although both BBc6R8 and Ter331 stimulated branching density and angle, no gene was found to be commonly regulated within these two strains. This incongruence between macroscopic observation and gene regulation could be explained either by the existence of distinct molecular pathways involved in hyphal branching or by earlier transcriptomic responses not included in this study. In contrast, the respective stimulation and inhibition of the radial growth of the fungus by strain BBc6R8 and Ter331 correlated with the respective increase and decrease in gene transcription linked to primary metabolism such as protein synthesis, transcription regulation and DNA replication (Fig. 2). These opposing transcriptional responses could therefore signify that regulation of the fungal metabolic activity is linked to its growth rate. Indeed an increase of cell growth requires elevated metabolic activity, while growth arrest in the presence of Ter331 would lead to a distinct decrease of metabolic activity. Such repression of the primary metabolism and growth in response to stressful conditions and starvation has been also described in Saccharomyces and Neurospora [25, 26].
Despite the high specificity of the fungal transcriptomic response to the three bacterial strains, four genes were found to be similarly regulated in response to the three bacterial strains. Therefore, we might conclude that some L. bicolor S238N pathways involved in the perception of bacteria could be commonly induced by all strains. Several earlier studies have suggested that some fungi could possibly have specific receptor systems for detecting other microorganisms [27, 28], although these receptors have yet to be discovered. The lipopolysaccharide binding-like protein encoding gene tectonin II is of particular interest. In other organisms, tectonins are able to bind bacteria through specific attachment to bacterial lipopolysaccharides [29, 30]. Preliminary data indicate that the protein is located on the cell wall of the fungus (data not shown) and that it is able also to agglutinate Gram-negative bacteria [31]. Physical adherence mediated by LPS and/or lectins has been observed in fungal–bacterial interaction [6, 32]. It would be tempting to speculate that this tectonin II is involved in physical interaction with bacteria and that it could be involved in the detection of bacteria. Further characterizations are currently being performed to determine its precise role in the physiology of the fungus and in the interaction with bacteria.
The three bacterial strains reacted differently to the presence of the ectomycorrhizal fungus with regulation of different sets of genes. Despite these differences, it should be noted that transcript regulations involved in nutrient acquisition and primary metabolism occurred in the three bacteria, suggesting the existence of trophic interactions between the bacteria and the fungus. In the presence of strain BBc6R8, this regulation mirrored the sixfold over-expression of a L. bicolor gene encoding a MFS transporter at the same stage of the interaction. Considered together, these data suggest that the two microorganisms modulate their nutrient absorption and metabolism in the presence of each other. Whether the fungus and the helper bacterium compete for or exchange nutrients remains to be determined. In addition, transcripts potentially involved in cell wall remodelling were regulated in the three bacteria, although different transcripts were regulated for each strain. It is therefore possible that bacteria modify their cell wall structure in response to the presence of the ectomycorrhizal fungus. Further studies will be necessary to determine whether bacteria remodel their cell walls in response to the presence of the fungus and what the role is of this remodelling in the output of the interaction. Some interactions between fungi and bacteria only occur when the two partners have close physical interaction [6]. For example, the actinomycete Streptomyces hygroscopicus only produces orsellinic acid when it is adhering to hyphae of A. nidulans [24].
Strain Ter331 and Pf29Arp have been shown to react at the transcriptomic level during their interaction with the ascomycete A. niger [19] and the wheat pathogen Gaeumannomyces graminis [12, 20, 33, 34], respectively. Interestingly, both strains reacted quite differently when challenged with L. bicolor S238N. In contrast to the interaction with A. niger during which Ter331 strongly responded at the transcriptomic level to the presence of A. niger (8 % of the transcriptome), almost no modification of the transcriptome of the bacterium was observed in the presence of L. bicolor S238N (0.2 % of the transcriptome). As such, Ter331 may not perceive the presence of L. bicolor S238N and would constitutively produce antagonistic molecule(s). This “invisibility” of L. bicolor could be due to its reduced production of oxalic acid [35] which was involved in the interaction between Ter331 towards A. niger [19]. Indeed while A. niger can produce up 70 mM of oxalic acid from 10 g of sugar [36], L. bicolor S238N only produces around 5 μM of oxalic acid from the same amount of sugar [35]. While the mycophagous strain Ter331 induced the same macroscopic phenotype in L. bicolor S238N and A. niger by inhibiting their radial growth and by stimulating their hyphal branching, the molecular mechanisms of the interaction are probably different. The A. niger/Ter331 interaction seems to be based on a ping-pong interplay in which both organisms fight back through the production of secondary metabolites and competition for nitrogen [19]. L. bicolor S238N may also defend itself against Ter331 through the production of antibacterial compounds as suggested by the overexpression of clitocypin-encoding genes. Clitocypins are cystein protease inhibitors with a broad inhibitory profile and which have been proposed to be involved in the defence against pathogens [37, 38]. This potential role in defence against pathogens is supported by the nematicide activity of L. bicolor clitocypins (Plett and Martin, personal communication). However, while the ping-pong between A. niger and Ter331 resulted in reduced growth of both microorganisms, only the growth of L. bicolor S238N was inhibited during the interaction with Ter331 (data not shown). This growth inhibition correlated with a decrease in the expression of many genes linked to the primary metabolism of the ectomycorrhizal fungus. Whether this shutdown reflects a stress response due to the presence of the antagonist bacterium or a proactive response of the fungus to the activities of the bacterium remains to be determined.
No transcripts were found to be commonly regulated in Pf29Arp during the interaction with the pathogenic fungus G. graminis [12] compared to L. bicolor S238N, suggesting a specific response. However, in both cases, transcripts involved in metabolic processes and cell envelop biogenesis were regulated. Interestingly, a common pattern of response involving cell envelop biogenesis, transcription regulation and metabolic processes was observed, although different sets of genes were involved in each bacterium during the interaction with the fungus. More particularly, regulation of metabolic processes appeared to be a common point of the four microorganisms, suggesting the importance of trophic interactions in driving the interactions in our setup. However, the type of trophic interaction most likely changes, depending on the pair of microorganisms considered, since various types of metabolic processes were regulated. We can therefore expect the occurrence of a complex network of trophic interactions in the microbial communities selected in the ectomycorrhizosphere. Global food webs between mycorrhizal fungi bacteria and microfauna have yet to be described in greater detail [39]; to date, our knowledge about the metabolites exchanged and consumed in situ by interacting microorganisms is limited.
Conclusion
This study provides a glimpse of molecular mechanisms involved in ectomycorrhizal interaction with different soil bacteria. Based on analysis of transcriptomic datasets, we may conclude that a high degree of specificity exists within these molecular pathways. Hence, the ectomycorrhizal fungus L. bicolor S238N appears to differentially adapt its primary metabolism in response to distinct bacterial strains, which in turn modify the primary metabolism of these bacterial strains. Whether these changes are due to an exchange of metabolism or simply a competition for nutrients remains to be determined. In contrast to these specific modifications observed, the physical interactions between the fungus and the bacterial strains rely on conserved molecular mechanisms linked to cell wall modifications. Finally, a high proportion of differentially regulated genes was found to encode proteins with unknown functions. As these genes could potentially play an important role in how microorganisms adapt to their environment, understanding how they function merits further investigation.
References
Bulgarelli D, Rott M, Schlaeppi K et al (2012) Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488:91–95
Lundberg DS, Lebeis SL, Paredes SH et al (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90. doi:10.1038/nature11237
Cho I, Blaser MJ (2012) The human microbiome: at the interface of health and disease. Nat Rev Genet 13:260–270
Berendsen RL, Pieterse CM, Bakker PA (2012) The rhizosphere microbiome and plant health. Trends Plant Sci 17:478–486
Smith S, Read D (2008) Mycorrhizal symbiosis, 3rd edn. Mycorrhizal symbiosis 800
Frey-Klett P, Burlinson P, Deveau A et al (2011) Bacterial–fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol Mol Biol Rev 75:583–609
Horan DP, Chilvers GA (1990) Chemotropism: the key to ectomycorrhizal formation? New Phytol 116:297–301
Deveau A, Palin B, Delaruelle C et al (2007) The mycorrhiza helper Pseudomonas fluorescens BBc6R8 has a specific priming effect on the growth, morphology and gene expression of the ectomycorrhizal fungus Laccaria bicolor S238N. New Phytol 175:743–755
Riedlinger J, Schrey SD, Tarkka MT et al (2006) Auxofuran, a novel metabolite that stimulates the growth of fly agaric, is produced by the mycorrhiza helper bacterium Streptomyces strain AcH 505. Appl Environ Microbiol 72:3550–3557. doi:10.1128/AEM.72.5.3550-3557.2006
Aspray TJ, Jones EE, Davies MW et al (2013) Increased hyphal branching and growth of ectomycorrhizal fungus Lactarius rufus by the helper bacterium Paenibacillus sp. Mycorrhiza 23:403–410
Schrey SD, Schellhammer M, Ecke M et al (2005) Mycorrhiza helper bacterium Streptomyces AcH 505 induces differential gene expression in the ectomycorrhizal fungus Amanita muscaria. New Phytol 168:205–216. doi:10.1111/j.1469-8137.2005.01518.x
Barret M, Frey-Klett P, Boutin M et al (2009) The plant pathogenic fungus Gaeumannomyces graminis var. tritici improves bacterial growth and triggers early gene regulations in the biocontrol strain Pseudomonas fluorescens Pf29Arp. New Phytol 181:435–447
Chapon A, Guillerm A-Y, Delalande L et al (2002) Dominant colonisation of wheat roots by Pseudomonas fluorescens Pf29A and selection of the indigenous microflora in the presence of the take-all fungus. Eur J Plant Pathol 108:449–459
Leveau JHJ, Uroz S, de Boer W (2010) The bacterial genus Collimonas: mycophagy, weathering and other adaptive solutions to life in oligotrophic soil environments. Environ Microbiol 12:281–292. doi:10.1111/j.1462-2920.2009.02010.x
Di Battista C, Selosse M-A, Bouchard D et al (1996) Variations in symbiotic efficiency, phenotypic charachters and ploidy level among different isolates of the ectomycorrhizal basidiomycete Laccaria bicolor strain S238. Mycol Res 100:1315–1324
Sambrook J, Fritsch E, Maniatis T (1989) Molecular cloning: a laboratory manual. 1626
Baldi P, Long AD (2001) A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17:509–519
Duplessis S, Courty PE, Tagu D, Martin F (2005) Transcript patterns associated with ectomycorrhiza development in Eucalyptus globulus and Pisolithus microcarpus. New Phytol 165:599–611
Mela F, Fritsche K, de Boer W et al (2011) Dual transcriptional profiling of a bacterial/fungal confrontation: Collimonas fungivorans versus Aspergillus niger. ISME J 5:1494–1504. doi:10.1038/ismej.2011.29
Barret M, Frey-Klett P, Guillerm-Erckelboudt AY et al (2009) Effect of wheat roots infected with the pathogenic fungus Gaeumannomyces graminis var. tritici on gene expression of the biocontrol bacterium Pseudomonas fluorescens Pf29Arp. Mol Plant Microbe Interact 22:1611–1623
Frey P, Frey-Klett P, Garbaye J et al (1997) Metabolic and genotypic fingerprinting of fluorescent Pseudomonads associated with the Douglas fir—Laccaria bicolor mycorrhizosphere. Appl Environ Microbiol 63:1852–1860
Uroz S, Courty PE, Pierrat JC et al (2013) Functional profiling and distribution of the forest soil bacterial communities along the soil mycorrhizosphere continuum. Microb Ecol 66:404–415. doi:10.1007/s00248-013-0199-y
Traxler MF, Watrous JD, Alexandrov T et al (2013) Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. mBio. doi:10.1128/mBio.00459-13
Schroeckh V, Scherlach K, Nützmann H-W et al (2009) Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci U S A 106:14558–14563. doi:10.1073/pnas.0901870106
Gasch AP, Werner-Washburne M (2002) The genomics of yeast responses to environmental stress and starvation. Funct Integr Genom 2:181–192. doi:10.1007/s10142-002-0058-2
Xie X, Wilkinson HH, Correa A et al (2004) Transcriptional response to glucose starvation and functional analysis of a glucose transporter of Neurospora crassa. Fungal Genet Biol 41:1104–1119. doi:10.1016/j.fgb.2004.08.009
Paoletti M, Saupe SJ (2009) Fungal incompatibility: evolutionary origin in pathogen defense? Bioessays 31:1201–1210. doi:10.1002/bies.200900085
Silar P (2012) Hyphal interference: self versus non self fungal recognition and hyphal death. In: Guenther W (ed) Biocommunication of fungi. Springer, New York, pp 155–170
Huh CG, Aldrich J, Mottahedeh J et al (1998) Cloning and characterization of Physarum polycephalum tectonins. Homologues of Limulus lectin L-6. J Biol Chem 273:6565–6574
Low DHP, Frecer V, Le Saux A et al (2010) Molecular interfaces of the galactose-binding protein tectonin domains in host–pathogen interaction. J Biol Chem 285:9898–9907. doi:10.1074/jbc.M109.059774
Wohlschlager T, Butschi A, Grassi P et al (2014) Methylated glycans as conserved targets of animal and fungal innate defense. Proc Natl Acad Sci USA. doi:10.1073/pnas.1401176111
Peng X, Sun J, Iserentant D et al (2001) Flocculation and coflocculation of bacteria by yeasts. Appl Microbiol Biotechnol 55:777–781
Marchi M, Boutin M, Gazengel K et al (2013) Genomic analysis of the biocontrol strain Pseudomonas fluorescens Pf29Arp with evidence of T3SS and T6SS gene expression on plant roots. Environ Microbiol Rep 5:393–403. doi:10.1111/1758-2229.12048
Daval S, Lebreton L, Gazengel K et al (2011) The biocontrol bacterium Pseudomonas fluorescens Pf29Arp strain affects the pathogenesis-related gene expression of the take-all fungus Gaeumannomyces graminis var. tritici on wheat roots. Mol Plant Pathol 12:839–854
Cumming J, Swiger T, Kurnik B, Panaccione D (2001) Organic acid exudation by Laccaria bicolor and Pisolithus tinctorius exposed to aluminum in vitro. Can J For Res 31:703–710
Ruijter GJG, van de Vondervoort PJI, Visser J (1999) Oxalic acid production by Aspergillus niger: an oxalate-non-producing mutant produces citric acid at pH 5 and in the presence of manganese. Microbiology 145:2569–2576
Sabotič J, Kos J (2012) Microbial and fungal protease inhibitors—current and potential applications. Appl Microbiol Biotechnol 93:1351–1375. doi:10.1007/s00253-011-3834-x
Sabotič J, Kilaru S, Budič M et al (2011) Protease inhibitors clitocypin and macrocypin are differentially expressed within basidiomycete fruiting bodies. Biochimie 93:1685–1693. doi:10.1016/j.biochi.2011.05.034
Eissfeller V, Beyer F, Valtanen K et al (2013) Incorporation of plant carbon and microbial nitrogen into the rhizosphere food web of beech and ash. Soil Biol Biochem 62:76–81
Acknowledgments
A. Deveau was supported by a scholarship from INRA, Lorraine Region and the Foreign Office exchange program Van Gogh. This work was supported by the French National Research Agency through the Laboratory of Excellence ARBRE (ANR-11-LABX-0002-01). We would like to thank A. Kohler for her help in submitting data to the GEO database and Aimee Orsini for proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Deveau, A., Barret, M., Diedhiou, A.G. et al. Pairwise Transcriptomic Analysis of the Interactions Between the Ectomycorrhizal Fungus Laccaria bicolor S238N and Three Beneficial, Neutral and Antagonistic Soil Bacteria. Microb Ecol 69, 146–159 (2015). https://doi.org/10.1007/s00248-014-0445-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-014-0445-y