
Abstract
Fungus-growing termites, their symbiotic fungi, and microbiota inhibiting their intestinal tract comprise a highly efficient cellulose-hydrolyzing system; however, little is known about the role of gut microbiota in this system. Twelve fosmid clones with β-glucosidase activity were previously obtained by functionally screening a metagenomic library of a fungus-growing termite, Macrotermes annandalei. Ten contigs containing putative β-glucosidase genes (bgl1–10) were assembled by sequencing data of these fosmid clones. All these contigs were binned to Bacteroidetes, and all these β-glucosidase genes were phylogenetically closed to those from Bacteroides or Dysgonomonas. Six out of 10 β-glucosidase genes had predicted signal peptides, indicating a transmembrane capability of these enzymes to mediate cellulose hydrolysis within the gut of the termites. To confirm the activities of these β-glucosidase genes, three genes (bgl5, bgl7, and bgl9) were successfully expressed and purified. The optimal temperature and pH of these enzymes largely resembled the environment of the host’s gut. The gut microbiota composition of the fungus-growing termite was also determined by 454 pyrosequencing, showing that Bacteroidetes was the most dominant phylum. The diversity and the enzyme properties of β-glucosidases revealed in this study suggested that Bacteroidetes as the major member in fungus-growing termites contributed to cello-oligomer degradation in cellulose-hydrolyzing process and represented a rich source for β-glucosidase genes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Termites comprise one of the most efficient lignocellulose conversion biosystems on earth, and it has been found that they do so with major help from their symbiotic microorganisms [1, 2]. In lower termites, protists play major roles in plant matter degradation, while in higher termites, bacteria are proved to be largely responsible [3]. Among higher termites, Macrotermitinae spp., abundant and influential in Asia and Africa, are the most distinctive, as they obligately cultivate a basidiomycete fungi of genus Termitomyces in their nests [4], earning the name of fungus-growing termites. The symbiotic relationship between fungus-growing termites and the Termitomyces fungi is known to be maintained by a most complex polyethism implemented by different castes and ages [5]. Briefly, old workers forage for plant matters and carry them back to the nest, where young workers ingest the collected plant matters and produce largely undigested primary feces to construct a fungus comb (or a fungus garden) as consortium for growth of their ectosymbiotic fungi. As the fungus grows, in all forms of mycelium, fungus nodules (aggregate of asexual spores of Termitomyces), and seasonally flourished fruit bodies, young workers ingest fungus nodules while old workers feed on the aged part of fungus combs which is composed mainly of digested plant matters and mature mycelium. Meanwhile, as other higher termites, these termites also harbor a diverse milieu of gut bacteria whose compositions have been extensively studied [5–9].
In macrotermitine termites, the plant lignocellulose degradation is carried out within a multipartite symbiosis involving the termite host, the gut microbes, and the ectosymbiotic fungi Termitomyces [10, 11]. Endogenous cellulases from macrotermitine termite have been reported for both endoglucanase expressed in the salivary glands of the fungus grower Odontotermes formosanus [12] and β-glucosidases expressed primarily in the midgut of the fungus grower Macrotermes barneyi [13], suggesting that termite hosts themselves directly take part in the hydrolysis of cellulose. The symbiotic fungi could help the host to degrade lignin [14, 15], in addition to provision of cellulase and hemicellulase [16–18], while symbiotic roles of gut microbes residing in fungus-growing termite, especially in the process of lignocellulose degradation, remained largely undefined until several of recent works [19–21].
Gut microbiota of the fungus grower Odontotermes yunnanensis has been investigated by 454 pyrosequencing-based overall metagenomic analysis [20]. Carbohydrate-active enzymes (CAZymes) harbored by this microbiome, which are distributed in many protein families, are related to the hydrolysis of cellulose and hemicellulose [20]. This study suggested that the intestinal bacteria of fungus-growing termites mainly participate in the hydrolysis of cellulose and hemicellulose, but not in the degradation of lignin. Most of these CAZymes belonged to catalogs of debranching enzymes or oligosaccharide-processing enzymes; however, their properties, including substrates specificity, enzyme activity, optimal pH, and temperature, have not been characterized further. Functional screening of fosmid metagenome libraries of other fungus growers also indicated that hemicellulase, including arabinofuranosidase (debranching enzymes), xylosidase (oligosaccharide-processing enzyme), and xylanase, are present in the microbiome of the gut of fungus-growing termites, and characterization of these enzymes suggested that gut microbiota participated in hydrolysis of hemicellulose [19, 21].
In our previous study, a fosmid library was constructed from gut metagenome of old adult workers of the fungus-growing termite Macrotermes annandalei, and 12 clones with β-glucosidase (oligosaccharide-processing enzyme) activity were recovered from functional screening of 10,000 clones, which suggested that gut microbiota also took part in the hydrolysis of cellulose, especially in cello-oligomer degradation [19]. In this study, β-glucosidase genes were predicted from sequences of these fosmids and heterologously expressed and characterized, which provided more information about the role of gut microbiota in cellulose hydrolysis. The composition of gut microbiota was also investigated by 16S rRNA gene survey, and the correlations between the symbiotic bacteria and their hosts were attempted.
Materials and Methods
Chemicals and Reagents
Restriction enzymes and T4 DNA ligase used for cloning were obtained from Takara (Dalian, China). The Plasmid MiniPrep Kit for plasmid purification and DNA Gel Extraction Kit for DNA purification were obtained from Axygen (Hangzhou, China). All other reagents were of analytical grade and were purchased from Sangon (Shanghai, China).
Termite Collection and Gut Bacterial Genomic DNA Extraction
Termite sampling and DNA extraction have been done and described in the previous study [19], in which whole guts of about 2,500 old adult workers in a nest of M. annandalei were used for metagenomic DNA extraction, and the extracted DNA was used directly as the template for amplifying the V3 regions of the 16S rRNA genes.
Vectors and Strains
EPI300-T1R (Epicentre) was used for fosmid library construction. The selected β-glucosidase gene was cloned into plasmid pET-22b(+), and the expression was carried out in Escherichia coli BL21 (Novagen).
Microbial Diversity Survey
In order to investigate the microbial composition in the given termite gut, V3 regions of bacterial 16S rRNA genes were amplified and subjected to 454 pyrosequencing. Original metagenomic DNA was used directly as the template for 16S rRNA gene amplification. The forward primer was 341f,5′-ACGTCACCTACGGGAGGCAGCAG-3′, and the reverse primer was 534r,5′-ACGTCAATTACCGCGGCTGCTGG-3′. The six bases underlined in the primers were unique barcode for distinguishing PCR product from other samples. Reaction conditions were described as the follows: 25 μl of PCR reaction mixture, which contained 1 U of Ex Taq polymerase, 2.5 μl of the corresponding 10× amplification buffer, 200 mmol/l of each deoxynucleoside triphosphate (dNTP), and 20 pmol of each primer, and 10 ng of total genomic DNA. PCR reactions were performed using the following program: 2 min denaturation at 95 °C followed by 24 cycles of 1 min at 94 °C (denaturation), 1 min for annealing (0.5 °C reduced for every cycle from 65 to 56 °C followed by 5 cycles at 55 °C), and 1 min at 72 °C (elongation), with a final extension at 72 °C for 6 min. Then, the PCR product was subjected to 454 pyrosequencing (GS FLX sequencing system). All sequences were aligned using the NAST aligner [22] from the Greengenes [23] website (http://greengenes.lbl.gov/). All possible chimeras were detected with Bellerophon [24] (version 3) hosted at Greengenes and then excluded from subsequent analyses. All the aligned sequences were inputted to ABR software to calculate distance matrix, and then, the distance matrices were used to calculate operational taxonomic units (OTUs) (similarity cutoff value 97 %) using DOTUR software [25]. The phylogeny of each read was determined by RDP Classifier software, and 50 % confidence was chosen because of the short length of the reads (<250 bp) [26].
Assembly and Annotation of Sequencing Data of Fosmids
Fosmid DNAs of 12 β-glucosidase-positive clones were pooled and then sequenced by using 454 pyrosequencing (GS FLX sequencing system) as described previously [19]. The 454 reads were assembled into different contigs by Newbler software (http://www.454.com). MEGAN software (http://ab.inf.uni-turbingen.de/software/megan/) was employed to assign the taxonomy of ORFs in each contig [21, 27, 28]. If 50 % ORFs of a contig were all binned to the same phylum, this contigs could be assigned to this phylum [21]. The ORFs of contigs were predicted by FgenesB (Softberry, Goteborg, Sweden). β-Glucosidases were annotated from these ORFs by using BLASTX against the NCBI nonredundant protein database (http://www.ncbi.nlm.nih.gov). Annotation of the other conserved domains was performed using Pfam (http://pfam.sanger.ac.uk/). Predication of signal peptides of these β-glucosidases was performed by SignalP 4.1 in CBS (http://www.cbs.dtu.dk/services/SignalP/). Molecular mass and isoelectric point were predicted by ExPASy (http://www.expasy.ch/tools/protparam.html). β-Glucosidase genes annotated from the contigs were aligned with their nearest neighbors using MAFFT version 6.864 [29], and phylogenetic analysis was performed with MEGA 5.0. Distance was calculated based on the maximum likelihood criterion. The bootstrap confidence values were obtained based on 100 replicates. The sequences of β-glucosidase genes were deposited in GenBank, and the accession numbers are JN848956, JN848957, JN848959, and JN848961 to JN848967. The accession numbers of the contigs are KJ095701–KJ095710.
Expression of β-Glucosidase Genes and Purification
In order to express β-glucosidase, the β-glucosidase genes were amplified from the fosmid DNAs by 35 cycles of PCR using the primer (Table 1). The amplicon was cloned into a pET22b(+) expression vector and then transformed into E. coli BL21. The bacteria were cultured at 37 °C in LB medium containing ampicillin (50 μg ml−1) to an optical density of 0.6, and gene expression was induced by the addition of 50 μM l−1 isopropyl-β-d-thiogalactopyranoside (IPTG) at 28 °C for 16 h. Cells were harvested by centrifugation, and the cell pellet was resuspended in 2.5 ml lysis buffer (50 mM l−1 NaH2PO4, 300 mM l−1 NaCl, 10 mM l−1 imidazole, pH 8.0) and then disrupted by ultrasonic. The recombinant protein was purified using a nickel-nitrilotriacetic acid (NTA) column (Qiagen). The protein concentration was determined with a DC Protein Assay Kit (Bio-Rad Laboratories), and the target protein was detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie Blue G-250.
β-Glucosidase Activity Assay
β-Glucosidase activity was assayed at the optimum temperature by incubating 0.4 μg of diluted enzyme with 2.5 mM l−1 p-nitrophenyl-β-d-glucoside (Sigma) in 50 mM l−1 Na-phosphate buffer (pH 6.5) for 5 min, in triplicate. The reaction was terminated by adding 200 μl of 1 M l−1 NaCO3. The release of p-nitrophenol (pNP) was determined by measuring the absorbance at 405 nm using a Multiskan Spectrum spectrophotometer (Thermo Scientific, Finland). One unit of β-glucosidase activity was defined as the amount of enzyme required to hydrolyze 1 μmol of pNP-β-d-glucoside per minute under the standard assay conditions stated above.
To determine the optimum temperature, the standard activity assay was performed between 20 and 75 °C in 50mM l−1 Na-phosphate buffer (pH7.5). The optimum pH of three β-glucosidases was measured at the optimum temperature in the pH range of 4.5–5 (50 mM l−1 Na-acetate buffer) and 5.5–8.5 (50 mM l−1 Na-phosphate buffer).
Substrate specificity of BGL5 was determined by replacing the p-nitrophenyl-β-d-glucoside with pNP-β-d-cellobioside, cellobiose, and salicin. For 5 min, 2.5 % (w/v) substrates were incubated with 0.4 μg purified protein at 30 °C, pH 6.5. The enzyme assay for pNPC and salicin was under the experimental conditions described above. The enzyme activity for cellobiose degradation was assayed according to the Glucose Assay Kit (GAGO20, Sigma) manual.
The kinetic characteristics of BGL5 were measured with p-nitrophenyl-β-d-glucoside at concentrations ranging from 0 to 10 mM l−1, and K m and V max were calculated based on the Lineweaver-Burk method.
To determine the relationship of various metal ions and the enzyme activity, 50 mM l−1 TMEMD-HCl buffer containing 1 mM metal ions was incubated with the purified protein. The enzyme activity was assayed as described above.
Results
Bacterial Diversity
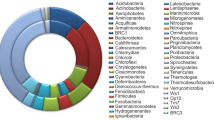
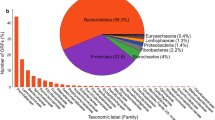
As the limitations existed when culture-based method was employed to study the intestinal bacteria, 454 pyrosequencing was used to characterize the bacterial composition in the termites’ gut. At 97 % cutoff, 1,185 qualified 16S rRNA gene sequences were obtained and classified into 378 OTUs. These 16S rRNA genes were mainly distributed into nine phyla. The relative abundance of each phylum was calculated, and the results indicated that the sequences affiliated to Bacteroidetes constituted the most dominant phylum in this termite (73.2 %), followed by Firmicutes (18.1 %) (Fig. 1a).

Bacterial abundance in termites’ gut. a Pyrosequencing of bacterial 16S rRNA gene V3 region in termites’ gut was performed, and the sequence was divided into different groups based on distance matrix. b The distribution and proportion of genus in Bacteroidales
Bacteroidetes was the most abundant phylum in termites’ gut, and sequences affiliated with Bacteroidetes were further analyzed. There were 872 sequences affiliated to Bacteroidetes, including Flavobacteria (1.4 %), Sphingobacteria (0.1 %), Bacteroidia (51.6 %), and unclassified Bacteroidetes(46.9 %). Within Bacteroidia, Dysgonomonas was the most dominant genus observed in the termites gut, and the proportion of this genus in Bacteroidia was 25.3 % (Fig. 1b).
Taxonomy Binning of Fosmid Contigs Containing β-Glucosidase Genes
By Newbler software, 454 reads of 12 fosmid clones with β-glucosidase activity were assembled into 55 large contigs (>500 bp) and 10 contigs were predicted to encode β-glucosidase genes. In order to reveal taxonomy source of these β-glucosidase genes, MEGAN program was used to bin all the 10 contigs containing β-glucosidase genes. The results indicated that all the contigs of β-glucosidases in this study can be assigned to the phylum Bacteroidetes (Bacteroidales, Dysgonomonas, etc.) (Table 2), suggesting that this phylum was important in affording functional enzyme within termites gut.
Phylogenetic Analysis of β-Glucosidase Genes
Based on the sequence analysis, all these β-glucosidases belong to GH3 family. Phylogenetic analyses based on amino acids were performed, and the results were shown in Fig. 2. BGL5 formed deep-branched lineages compared to other members analyzed. BGL5 was closely related to one β-glucosidase isolated from Bacteroides sp. (56 % amino acid identity). BGL2, BGL6, BGL7, BGL8, and BGL9 showed a high similarity to the hypothetical protein from the genus Dysgonomonas (amino acid identity varied from 81 to 100 %). BGL1, BGL3, BGL4, and BGL10 had a high homology to the enzymes originated from the genus Bacteroides (amino acid identity varied from 58 to 77 %).

Phylogenetic analysis of β-glucosidase detected in the termites’ gut. The protein sequence of BGL5 was aligned with the reference sequences retrieved from GenBank. The phylogenetic analysis was inferred by maximum likelihood analysis using amino acid sequence. The reference sequences are marked with source strains and GenBank accession numbers are in parentheses
Functional Characterization of β-Glucosidases
According to SignalP prediction, 6 out of 10 β-glucosidase genes possess the N-terminal signal peptides indicating a transmembrane capability of the enzymes (Fig. 3), suggesting that these enzymes could function extracellularly to take part in cellulose hydrolysis. The structures of these genes were predicted, and the results indicated that in addition to GH3 domains, eight of them also possessed Fn3-like domain at C-terminal (Fig. 3).

Domain architecture of the β-glucosidase genes. GH glycoside hydrolase. The asterisk indicated the successfully purified proteins
All of these genes were tried to be expressed in E. coli; however, only three of them (bgl5, bgl7, and bgl9) were successfully expressed. bgl5 is composed of 2,214 bp, encoding a protein of 738 amino acids, with a predicted molecular mass of 79.4 kDa and a predicted isoelectric point of 5.43. bgl7 encoded 798 amino acids, with a predicted molecular mass of 85.9 kDa and a predicted isoelectric point of 5.51.bgl9 encoded 797 amino acids, and the predicted molecular mass and predicted isoelectric point were 86.2 kDa and 5.99, respectively.
All these genes were cloned in the pET22b(+) expression vectors and expressed in E. coli BL21. The protein was detected in both the supernatant and precipitation after IPTG induction. With an N-terminal 6× His tag, BGL5, BGL7, and BGL9 were successfully purified by an elution buffer containing 250 mM l−1 imidazole (Supplementary Fig. 1). The molecular mass estimated by SDS-PAGE analysis agreed with the predicated values. The optimum pH of these enzymes was 6.5, and the optimum temperature of BGL5 and BGL9 was 30–35 °C. The optimum temperature of the other enzyme BGL7 was 50 °C. The enzyme activity was detected with p-nitrophenyl-β-d-glucoside as the substrate under the optimum pH and temperature of each enzyme, and the results indicated that BGL5 showed the strongest catalytic activity (Table 3).
Considering the similar structure and higher enzyme activity, the substrate specificity of BGL5 was further characterized. BGL5 hydrolyzed p-nitrophenyl-β-d-glucoside (pNPG) with an activity of 42.3 ± 2.0 U mg−1, whereas it hydrolyzed p-nitrophenyl cellobioside (pNPC) and cellobiose at a rate of 16.85 ± 0.53 and 5.26 ± 0.37 U mg−1, which were only 38 and 12 % of the rate to hydrolyze pNPG, respectively. Furthermore, it showed a very weak activity (1.21 ± 0.18 U mg−1) on salicin. These results indicated that BGL5 was a specific β-glucosidase and its substrate range was very limited. The V max and K m of BGL5 were determined based on a Lineweaver-Burk plot, which were 0.13 μmol min−1 mg−1 protein and 1.99 mg ml−1, respectively.
The effects of different metal ions and chemical reagents on BGL5 were also detected. It was shown that the enzymatic activity was significantly enhanced by Mg2+, Co2+, Zn2+, Fe3+, and Ni2+. BGL5 was inhibited by Ag+, Cu2+, and EDTA which reduced its enzymatic activity to 4.21, 18.11, and 72.81 %, respectively, of that without addition these items in the reaction (Fig. 4).

Effect of different metal ions on BGL5 activity. PBS was used as the control. All the experiments were conducted in triplicate, and the relative activity was shown in this figure
Discussion
The microbial communities harbored in the gut of fungus-growing termites have been effectively studied, and the dominance of Bacteroidetes and Firmicutes revealed here in M. annandalei largely resembles that revealed in other investigated fungus growers, including Macrotermes [5, 30], Odontotermes [8, 11, 20], and Pseudocanthotermes [21], and this differs obviously to the predominance of Spirochaetes in the revealed wood feeders [7, 31, 32], further indicating a diet-driven phylum-level divergence between gut microbiota in the fungus-growing and wood-feeding higher termites.
Compared to the well-established contributions of the symbiotic fungi for their hosts’ plant material decomposition [15, 17], the exploration of the function of intestinal bacteria in cellulose hydrolysis for their host appears particularly necessary and impendent. Here, extending the previous work [19], we further sequenced these β-glucosidase-positive clones and got 10 β-glucosidases genes. However, as expressing the target genes in a foreign host is the first step of functional screening, codon bias, missing substrates, and the inability to recognize foreign regulatory elements are several possibilities to limit the entirely identification of the potential genes or gene clusters [33], and more β-glucosidase genes may exist in termites’ guts. Taxonomy analysis showed that all the contigs were assigned to the phylum Bacteroidetes, and phylogenetic analysis indicated that most of the β-glucosidases showed a high identity to those from Dysgonomonas spp. or Bacteroides spp. 454 pyrosequencing of 16S rRNA gene indicated that Bacteroidetes was predominant in the gut microbiota. Members of the phylum Bacteroidetes employ a highly specialized system for polysaccharide utilization which involves a core set of polysaccharide-binding proteins, outer membrane transporters, and glycolytic enzymes to cleave large polysaccharides into fragments [34]. It has been suggested that Bacteroidetes play an important role in the cellulose or hemicellulose degradation in human gut or rumen [34–36]. Our study suggested that Bacteroidetes possess β-glucosidases genes, which suggested that in termites’ gut, Bacteroidetes as the dominant member played an important role in the last step of cellulose degradation.
There are 113 sequences (9.5 %) affiliated to Dysgonomonas spp. in this work. Dysgonomonas has been isolated from human sources and soil and showed positive reaction for β-glucosidases [33, 37–42]. However, the role of this genus in termite symbiotic system still remains elusive. In this study, 12 positive clones with β-glucosidase activity were screened from the termite gut fosmid library with 10,000 clones, and the hit rate was about 0.12 %. The hit rates for β-glucosidase of this fosmid library were modest compared with those of other metagenome libraries constructed from termite gut, biogas digest, and soils from wetland [21, 43].
Being a higher termite, M. annandalei lacks the flagellated protists, so the cellulolytic degradation process mainly depended on the gut microbes, symbiotic fungi, and host cellulases. β-Glucosidase activity in termites’ midgut (including the host tissue and the content) was higher than that in a fungus-growing termite hindgut or in the fungus nodules [44]. The expressed sequence tag (EST) analysis indicated that the symbiotic fungi in Macrotermes spp. were more active in the hydrolysis of backbones of cellulose, hemicellulose, and pectins than in the hydrolysis of oligomers [16]. Specifically, the proportion of β-glucosidase was much lower than that of endoglucanase and exoglucanase [16], indicating a potential limit of the symbiotic fungi in processing downstream cellobiose and cello-oligosaccharides, while the diverse and function of β-glucosidase genes revealed in this study indicate that gut microbiota may work cooperatively with the Termitomyces fungi by further dissimilating fungi-processed plant derivatives, including cellobiose and cellodextrin, for host’s plant matter degradation. This hypothesis is in accordance with our recent metagenomic overview of the degradative capacities of gut microbiome of another fungus-growing termite O. yunnanensis [20]. These findings will greatly broaden our understanding of the degradative symbiosis within fungus-growing termites.
Among the three major types of cellulases, β-glucosidases can hydrolyze cellobiose or cello-oligosaccharides to produce glucose, thus catalyzing the last and most critical step in cellulose degradation [45]. Cellobiose as an intermediate is known to exert a feedback inhibition on upstream cellulases; the timely removal of cellobiose by β-glucosidases is known to be able to accelerate the whole process of cellulose hydrolysis by reducing this feedback inhibition effect [46, 47]. The relative rates of hydrolysis of various substrates by the BGL5 were measured. β-Glucosidase mainly hydrolyze β-1,4-glucosidic bonds in cellobiose, short-chain cello-oligosaccharides and aryl-β-d-glucosides. Compared to other bacterial β-glucosidases retrieved by the metagenomic analyses from other environments, our enzymes had a relatively higher efficiency to hydrolyze pNPG [48, 49]. Eight out of the 10 β-glucosidases genes identified in this fungus-growing termite turn to include an Fn3-like domain at C-terminal, while the three enzymes expressed in our study all harbored the Fn3-like domain. It has been considered that this domain is helpful to improve the cellulose hydrolysis efficiency [50]; however, the function of Fn3-like domain in these enzymes has not been explored in this study.
This study revealed the diversity and enzyme property of β-glucosidase genes presented in the intestinal bacteria of fungus-growing termites, which highlighted the importance of intestinal bacteria in host’s cellulose degradation and their potential as a good resource for mining novel β-glucosidase genes. Meanwhile, the successful recovery of a batch of enzymes from fungus-growing termite gut metagenome features the advantages of metagenomics in its application in biotechnology [21, 32, 49, 51].
References
Breznak JA (1994) Role of microorganisms in the digestion of lignocellulose by termites. Annu Rev Entomol 39:453–487
Abo-Khatwa AN (1989) Termitomyces: a new source of potent cellulases. JKing Abulaziz Univ Science 1:51–59
Breznak JA (1982) Intestinal microbiota of termites and other xylophagous insects. Annu Rev Microbiol 36:323–343
Wood TG, Thomas RJ (1989) The mutualistic association between Macrotermitinae and Termitomyces. In: Wilding N, Collins NM, Hammond PM, Webber JF (eds) Insect-fungus interaction. Academic, London, pp 69–92
Hongoh Y, Ekpornprasit L, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Noparatnaraporn N, Kudo T (2006) Intracolony variation of bacterial gut microbiota among castes and ages in the fungus-growing termite Macrotermes gilvus. Mol Ecol 15(2):505–516
Mackenzie LM, Muigai AT, Osir EO, Lwande W, Keller M, Yoledo G, Boga HI (2007) Bacterial diversity in the intestinal tract of the fungus-cultivating termite Macrotermes michaelseni (Sjostedt). African Journal of Biotechnology 6(6):658–667
Hongoh Y, Deevong P, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Vongkaluang C, Noparatnaraporn N, Kudo T (2005) Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl Environ Microbiol 71(11):6590–6599
Shinzato N, Muramatsu M, Matsui T, Watanabe Y (2007) Phylogenetic analysis of the gut bacterial microflora of the fungus-growing termite Odontotermes formosanus. Biosci Biotechnol Biochem 71(4):906–915
Long YH, Xie L, Liu N, Yan X, Li MH, Fan MZ, Wang QA (2010) Comparison of gut-associated and nest-associated microbial communities of a fungus-growing termite (Odontotermes yunnanensis). Insect Sci 17(3):265–276
Rouland-Lefevre C, Inoue T, Johjima T (2006) Termitomyces/termite interactions. In: Konig H, Varma A (eds) Intestinal microorganisms of soil invertebrates. Springer, Berlin, pp 335–350
Mathew GM, Ju YM, Lai CY, Mathew DC, Huang CC (2012) Microbial community analysis in the termite gut and fungus comb of Odontotermes formosanus: the implication of Bacillus as mutualists. FEMS Microbiol Ecol 79(2):504–517
Tokuda G, Lo N, Watanabe H, Arakawa G, Matsumoto T, Noda H (2004) Major alteration of the expression site of endogenous cellulases in members of an apical termite lineage. Molecular Ecology 13(10):3219–3228
Wu Y, Chi S, Yun C, Shen Y, Tokuda G, Ni J (2012) Molecular cloning and characterization of an endogenous digestive beta-glucosidase from the midgut of the fungus-growing termite Macrotermes barneyi. Insect Mol Biol 21(6):604–614
Hyodo F, Inoue T, Azuma JI, Tayasu I, Tabe T (2000) Role of the mutualistic fungus in lignin degradation in the fungus-growing termite Macrotermes gilvus (Isoptera; Macrotermitinae). Soil Biol Biochem 32:653–658
Hyodo F, Tayasu I, Inoue T, Azuma JI, Kudo T, Abe T (2003) Differential role of symbiotic fungi in lignin degradation and food provision for fungus-growing termites (Macrotermitinae: Isoptera). Functional Ecology 17:186–193
Johjima T, Taprab Y, Noparatnaraporn N, Kudo T, Ohkuma M (2006) Large-scale identification of transcripts expressed in a symbiotic fungus (Termitomyces) during plant biomass degradation. Appl Microbiol Biotechnol 73(1):195–203
Martin M, Martin J (1978) Cellulose digestion in the midgut of the fungus-growing termite Macrotermes natalensis: the role of acquired digestive enzyme. Science 199:1453–1455
Matoub M, Rouland C (1995) Purification and properties of the xylanases from the termite Macrotermes bellicosus and its symbiotic fungus Termitomyces sp. Comp Biochem Physiol B Biochem Mol Biol 112(4):629–635
Liu N, Yan X, Zhang M, Xie L, Wang Q, Huang Y, Zhou X, Wang S, Zhou Z (2011) Microbiome of fungus-growing termites: a new reservoir for lignocellulase genes. Appl Environ Microbiol 77(1):48–56
Liu N, Zhang L, Zhou H, Zhang M, Yan X, Wang Q, Long Y, Xie L, Wang S, Huang Y, Zhou Z (2013) Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite (Odontotermes yunnanensis). PLoS One 8(7):e69184
Bastien G, Arnal G, Bozonnet A, Laguerre S, Ferreir F, GFaure R, Henrissat B, Lefevre G, Robe P, Bouchez O, Noirot C (2013) Mining for hemicellulases in the fungus-growing termite Pseudacanthotermes militaris using functional metagenomics. Biotechnol Biofuels 6:78. doi:10.1186/1754-6834-1186-1178
DeSantis TZ, Jr., Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34(Web Server issue):W394-399
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072
Huber T, Faulkner G, Hugenholtz P (2004) Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20(14):2317–2319
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71(3):1501–1506
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Huson DH, Auch AF, Qi J, Schuster SC (2007) Megan analysis of metagenomic data. Genome Res 17:377–386
Mitra S, Klar B, Huson DH (2009) Visual and statistical comparison of metagenomes. Bioinformatics 25:1849
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinformatics 9:286–298
Mackenzie LM, Muigai AT, Osir EO, Lwande W, Keller M, Toledo G, Hi B (2007) Bacterial diversity in the intestinal tract of the fungus-cultivating termite Macrotermes michaelseni (Sjostedt). African Journal of Biotechnology 6:658–667
Kohler T, Dietrich C, Scheffrahn RH, Brune A (2012) High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl Environ Microbiol 78(13):4691–4701
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernandez M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450(7169):560–565
Lawson PA, Falsen E, Inganas E, Weyant RS, Collins MD (2002) Dysgonomonas mossii sp. nov., from human sources. Syst Appl Microbiol 25(2):194–197
Martens EC, Koropatkin NM, Smith TJ, Gordon JI (2009) Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284(37):24673–24677
Dodd D, Mackie RI, Cann IK (2011) Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol 79(2):292–304
Mackenzie AK, Pope PB, Pedersen HL, Gupta R, Morrison M, Willats WG, Eijsink VG (2012) Two SusD-like proteins encoded within a polysaccharide utilization locus of an uncultured ruminant Bacteroidetes phylotype bind strongly to cellulose. Appl Environ Microbiol 78(16):5935–5937
Carpenter KJ, Keeling PJ (2007) Morphology and phylogenetic position of Eucomonympha imla (Parabasalia: Hypermastigida). J Eukaryot Microbiol 54(4):325–332
Hansen PS, Jensen TG, Gahrn-Hansen B (2005) Dysgonomonas capnocytophagoides bacteraemia in a neutropenic patient treated for acute myeloid leukaemia. APMIS 113(3):229–231
Hironaga M, Yamane K, Inaba M, Haga Y, Arakawa Y (2008) Characterization and antimicrobial susceptibility of Dysgonomonas capnocytophagoides isolated from human blood sample. Jpn J Infect Dis 61(3):212–213
Hofstad T, Olsen I, Eribe ER, Falsen E, Collins MD, Lawson PA (2000) Dysgonomonas gen. nov. to accommodate Dysgonomonas gadei sp. nov., an organism isolated from a human gall bladder, and Dysgonomonas capnocytophagoides (formerly CDC group DF-3). Int J Syst Evol Microbiol 50(6):2189–2195
Lawson PA, Carlson P, Wernersson S, Moore ER, Falsen E (2010) Dysgonomonas hofstadii sp. nov., isolated from a human clinical source. Anaerobe 16(2):161–164
Matsumoto T, Kawakami Y, Oana K, Honda T, Yamauchi K, Okimura Y, Shiohara M, Kasuga E (2006) First isolation of Dysgonomonas mossii from intestinal juice of a patient with pancreatic cancer. Arch Med Res 37(7):914–916
Collins MD, Lawson PA, Labrenz M, Tindall BJ, Weiss N, Hirsch P (2002) Nesterenkonia lacusekhoensis sp. nov., isolated from hypersaline Ekho Lake, East Antarctica, and emended description of the genus Nesterenkonia. Int J Syst Evol Microbiol 52(Pt 4):1145–1150
Martin MM, Matrtin JS (1979) The distribution and origins of the cellulolytic enzymes of the higher termite, Macrotermes natalensis. Physiological Zoology 52(1):11–21
Bhatia Y, Mishra S, Bisaria VS (2002) Microbial beta-glucosidases: cloning, properties, and applications. Crit Rev Biotechnol 22:375–407
Suto M, Tomita F (2001) Induction and catabolite repression mechanisms of cellulase in fungi. J Biosci Bioeng 92(4):305–311
Percival Zhang YH, Himmel ME, Mielenz JR (2006) Outlook for cellulase improvement: screening and selection strategies. Biotechnol Adv 24(5):452–481
Feng Y, Duan CJ, Liu L, Tang JL, Feng JX (2009) Properties of a metagenome-derived beta-glucosidase from the contents of rabbit cecum. Biosci Biotechnol Biochem 73(7):1470–1473
Jiang C, Ma G, Li S, Hu T, Che Z, Shen P, Yan B, Wu B (2009) Characterization of a novel beta-glucosidase-like activity from a soil metagenome. J Microbiol 47(5):542–548
Kataeva IA, Seidel RD 3rd, Shah A, West LT, Li XL, Ljungdahl LG (2002) The fibronectin type 3-like repeat from the Clostridium thermocellum cellobiohydrolase CbhA promotes hydrolysis of cellulose by modifying its surface. Appl Environ Microbiol 68(9):4292–4300
Brulc JM, Antonopoulos DA, Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE, White BA (2009) Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A 106(6):1948–1953
Acknowledgments
This work was supported by grants of the National Natural Science Foundation of China (31070098). We appreciate Professor Shubiao Wu from the University of New England for his critical reading and kind suggestions. We also give our thanks to Prashanth Singanallur and Lauren Christine Radlinski from the University of Illinois at Urbana-Champaign for proofreading. We appreciated Dr. Haokui Zhou from Department of Microbiology, The Chinese University of Hongkong and Lei zhang from Logic Informatics Co.,Ltd., for their kind help in data analysis.
Conflict of Interest
The authors have no conflict of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PPTX 48413 kb)
Rights and permissions
About this article
Cite this article
Zhang, M., Liu, N., Qian, C. et al. Phylogenetic and Functional Analysis of Gut Microbiota of a Fungus-Growing Higher Termite: Bacteroidetes from Higher Termites Are a Rich Source of β-Glucosidase Genes. Microb Ecol 68, 416–425 (2014). https://doi.org/10.1007/s00248-014-0388-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-014-0388-3