
Abstract
The phylum Fibrobacteres currently comprises one formal genus, Fibrobacter, and two cultured species, Fibrobacter succinogenes and Fibrobacter intestinalis, that are recognised as major bacterial degraders of lignocellulosic material in the herbivore gut. Historically, members of the genus Fibrobacter were thought to only occupy mammalian intestinal tracts. However, recent 16S rRNA gene-targeted molecular approaches have demonstrated that novel centres of variation within the genus Fibrobacter are present in landfill sites and freshwater lakes, and their relative abundance suggests a potential role for fibrobacters in cellulose degradation beyond the herbivore gut. Furthermore, a novel subphylum within the Fibrobacteres has been detected in the gut of wood-feeding termites, and proteomic analyses have confirmed their involvement in cellulose hydrolysis. The genome sequence of F. succinogenes rumen strain S85 has recently suggested that within this group of organisms a “third” way of attacking the most abundant form of organic carbon in the biosphere, cellulose, has evolved. This observation not only has evolutionary significance, but the superior efficiency of anaerobic cellulose hydrolysis by Fibrobacter spp., in comparison to other cellulolytic rumen bacteria that typically utilise membrane-bound enzyme complexes (cellulosomes), may be explained by this novel cellulase system. There are few bacterial phyla with potential functional importance for which there is such a paucity of phenotypic and functional data. In this review, we highlight current knowledge of the Fibrobacteres phylum, its taxonomy, phylogeny, ecology and potential as a source of novel glycosyl hydrolases of biotechnological importance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Genus Fibrobacter
Since Robert E. Hungate first isolated Fibrobacter succinogenes (formerly Bacteroides succinogenes) from the bovine rumen in 1947 [47, 48], members of the genus Fibrobacter have been considered to be major degraders of cellulosic plant biomass in the herbivore gut [49, 55, 114]. Fibrobacter is currently the sole formal genus of the bacterial phylum Fibrobacteres, which is phylogenetically related to the well-characterised Bacteroidetes and Chlorobi phyla [20, 71]. F. succinogenes was initially classified as B. succinogenes, and this was attributed to the historical broad genus definition for Bacteroides: “all anaerobic, Gram-negative, nonmotile or peritrichous, nonsporeforming rods that do not produce butyric acid from the fermentation of carbohydrates” [14]. However, this resulted in the accumulation of many unrelated species within the Bacteroides genus. It was suggested that, as B. succinogenes possessed mainly straight-chain fatty acids and lacked the membrane sphingolipids observed in other Bacteroides spp., it should be excluded from the genus [105].
Subsequently, 16S rRNA oligonucleotide cataloguing methods were used to demonstrate that B. succinogenes and Bacteroides amylophilus were in fact not closely related to the other Bacteroides species [98]. Montgomery and colleagues [91] utilised 16S rRNA gene sequencing methods to assess the phylogenetic relationship of B. succinogenes and its closest relatives, demonstrating that B. succinogenes isolates formed a phylogenetically coherent group, having no closely related organisms for which 16S rRNA gene sequence data were available. The genus Fibrobacter was circumscribed on this basis and contains only two recognised species, F. succinogenes and Fibrobacter intestinalis, both Gram-negative, obligate anaerobes that are the predominant bacterial colonisers and degraders of lignocellulosic plant material in the herbivore gut [91]. F. succinogenes comprised rumen isolates and F. intestinalis was the name assigned to the caecal isolates of B. succinogenes. Moreover, a previous study suggested that B. succinogenes isolates were sufficiently distant from other species to represent a distinct phylum [135]. Most recently, taxonomic distribution analysis of the predicted proteins in the F. succinogenes S85 genome confirmed that this species is indeed correctly classified at the phylum level [118].
Phenotypic Characteristics of Fibrobacter Isolates
Members of the genus Fibrobacter are defined as obligately anaerobic, non-sporeforming, Gram-negative, rods or pleiomorphic ovoid cells [91], 0.3 to 0.5 μm in diameter and 0.8 to 2.0 μm in length [48, 116]. The cells are able to migrate through agar medium by a mechanism comparable to that of Cytophaga spp. [48]. Fibrobacter spp. ferment xylan [36, 85, 109], glucose, cellobiose and cellulose, producing succinic and acetic acids, and sometimes a small amount of formic acid [91]. Ammonia [91], in addition to peptides and amino acids [4, 69], can be utilised as a source of nitrogen, and carbon dioxide, straight-chain and branched-chain fatty acids and one or more vitamins (typically biotin, p-aminobenzoic acid, B12 (cyanocobalamine) or thiamine) are also required for growth [91].
There are currently no definitive phenotypic characteristics that can be used to separate F. succinogenes and F. intestinalis. Previously, it was considered that F. succinogenes is a rumen bacterium while F. intestinalis inhabits the caecum [91]. This was later discredited when the use of rRNA gene-targeted oligonucleotide probes demonstrated that F. intestinalis is present in the rumen [113], and F. intestinalis strains LH1 and JG1 were subsequently isolated from the ovine rumen (Table 1). Furthermore, F. succinogenes was thought likely to be present in the intestine due to the carriage from rumen digesta [91], and this was confirmed by the isolation of stain GC5 from the bovine cecum (Table 1). Although it is evident that a loose relationship exists between the isolation site and the species, this cannot be used to definitively identify a Fibrobacter species [1]. The absolute requirement for biotin exhibited by F. succinogenes strains was the only known distinguishing phenotypic characteristic between the two species [49, 91]. However, it was subsequently found that two strains of F. intestinalis (LH1 and JG1) also require biotin for growth (Table 1) [1].
The Phylogeny of the Genus Fibrobacter
Despite the fact that there are currently no distinct phenotypic traits to distinguish F. succinogenes and F. intestinalis, there is considerable genetic distance between the two formally recognised species [1]. Furthermore, it has been suggested that the phylogenetic difference between them based on 16S rRNA gene sequence comparison is sufficient to designate them as belonging to two distinct genera [91] (Fig. 1). This is compounded by the fact that the evolutionary distance between F. succinogenes and F. intestinalis (as determined by 16S rRNA gene analysis) is similar to that between the bacterial genera containing Arthrobacter globiformis and Mycobacterium flavescens and deeper than that between Escherichia coli and Proteus vulgaris [91]. The diversity of Fibrobacter isolates was further characterised using comparative 16S rRNA gene sequencing and DNA/DNA hybridisations of a larger number of isolates (Table 1) [1]. Comparisons of the 16S rRNA gene of F. succinogenes and F. intestinalis demonstrated approximately 91% to 93% similarity, and genomic DNA similarity between the two species as determined by DNA/DNA hybridisation was less than 20% [1]. It is currently suggested that 20% DNA/DNA homology and approximately 95% 16S rRNA similarity [72] are the minimum allowable with a genus. Advances in next-generation sequencing technologies now make the application of comparative genomics a tangible approach for the ‘phylogenomic’ analysis of the Fibrobacteres phylum [137].

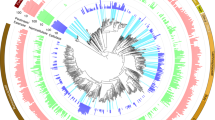
Phylogeny of the Fibrobacteres phylum. Maximum likelihood tree of 16S rRNA gene sequences belonging to the Fibrobacteres phylum. All sequences classified within the Fibrobacteres phylum and annotated as of ‘good’ quality were downloaded from the Ribosomal Database Project [19, 21] website in November 2010. Sequences were aligned using the MUSCLE aligner [30]. In order to compare the phylogeny of those sequences derived from environmental samples, termites and the herbivore gut, alignments were trimmed to include only sequences that contained positions corresponding to 153 to 1017 of the E. coli 16S rRNA gene. The remaining trimmed sequences were clustered into Operational Taxonomic Units (OTUs) at 95% similarity using CDHIT [46, 65]. A number of putative chimeric sequences were removed from the dataset after analysis with the Pintail chimera check program [3]. The representative sequences of each OTU (n = 42) were aligned using the Greengenes NAST aligner [29] and imported into Arb where the alignment was visually checked. A maximum likelihood tree was produced from the final alignment using PhyML online [37] with the HKY85 substitution model and the Shimodaira–Hasegawa-like approximate likelihood ratio test (aLRT) branch support method. Filled circles indicate nodes at which an aLRT value of >95% was observed, and unfilled circles denote nodes with aLRT values between 75 and 95%. Nucleotide sequence accession numbers for the representative sequence of each OTU are displayed on each node. The number of sequences clustering within each OTU is displayed in parentheses and numbered circles indicate the environmental niches represented within each OTU. Clusters highlighted in grey represent sequences that are affiliated with the two known cultivated species within the genus, F. succinogenes and F. intestinalis. The scale bar indicates 0.1 base substitutions per nucleotide
The study by Amann and colleagues [1] demonstrated four distinct lines of descent within the F. succinogenes lineage, designated F. succinogenes subsp. Succinogenes (subgroup 1) [91] and subgroups 2, 3 and 4 [1]. Of these, group 1 is considered to be the most important in cellulose degradation [55, 106, 107] due to its high metabolic activity and widespread presence on plant material. Koike et al. [57] detected only subgroups 1 and 3 in rumen digesta and on hay stems incubated in the rumen, with subgroup 1 dominating the Fibrobacter population on the less degradable hay stems. A study using fluorescence in situ hybridization (FISH) to determine the attachment of bacteria to hay within the rumen detected only F. succinogenes subgroups 1 and 2, with subgroup 1 cells representing the largest proportion of the Fibrobacter population on the stems [106]. Suppressive subtractive hybridization has been used to compare the genes of F. succinogenes S85 and F. intestinalis DR7, suggesting that 33% of F. intestinalis DR7 genes were specific to this strain [100] and 41% of F. succinogenes S85 genes were either absent from, or exhibited low similarity to, those of F. intestinalis DR7 [101]. However, as discussed above, there is little phenotypic difference between the two species and as such they remain within the same genus (Fig. 1). It is envisaged that a phylogenetically coherent family will be established for what is currently the genus Fibrobacter and its close relatives when more taxa are detected and identified.
Cellulose Degradation
Cellulose is the main structural component of higher plant cell walls and represents approximately 35–50% of plant dry weight [76]. It is also present in bacteria, fungi and some animals such as marine tunicates [96]. The process of photosynthesis creates extensive amounts of plant biomass and therefore cellulose, which must be degraded by cellulolytic microorganisms that are present in the soil, marine and lake sediments, water and animal guts. As such, one of the largest material flows in the biosphere is controlled by cellulolytic microorganisms [75]. Cellulose hydrolysis can occur under both aerobic and anaerobic conditions, with anaerobic hydrolysis accounting for 5% to 10% of global cellulose degradation [52, 127], which is substantial in view of the absolute amount of cellulosic biomass present in the environment. The physiological capability to degrade cellulose is distributed widely across the universal phylogenetic tree of life [75]. Within the Eubacteria, cellulose-degrading bacteria are largely concentrated in the aerobic order Actinomycetales (phylum Actinobacteria) and the anaerobic order Clostridiales (phylum Firmicutes). There is significant diversity in the physiology of cellulolytic bacteria, and on this basis they can be placed into three diverse physiological groups: (1) fermentative anaerobes, typically Gram-positive, such as Clostridium and Ruminococcus, but with a few Gram-negative species (Butyvibrio and Acetivibrio) that are phylogenetically related to the Clostridium assemblage (fibrobacters are within this group despite being phylogenetically unrelated); (2) aerobic Gram-positive bacteria, e.g. Cellulomonas and Thermobifida; and (3) aerobic gliding bacteria, such as Cytophaga and Sporocytophaga [75].
The majority of characterised cellulolytic microorganisms use either the free cellulase mechanism [133] in which multiple secreted enzymes act synergistically or complexes of cellulolytic enzymes bound to the outer cell wall (cellulosomes) [5] to digest cellulose (Fig. 2). Brown rot fungi are exceptional in their ability to attack cellulose using coupled oxidative enzymes [80]. For both the free cellulase mechanism most commonly used by aerobic organisms and the cellulosomes associated with anaerobic organisms, the β-1,4 linkages within the cellulose are hydrolysed by cellulases. The model of aerobic cellulose hydrolysis via the cell-free enzyme mechanism is based on the cellulase system of the aerobic fungus Trichoderma reesei and the ‘cellulosome’ mechanism of anaerobic bacteria and fungi (order Neocallimastigales) is based on the mechanisms of cellulolytic clostridia (reviewed by Lynd et al.) [74]. There are therefore substantial differences between the cellulose hydrolysis strategies employed by aerobic and anaerobic organisms [6]; the aerobic cell-free cellulase mechanism evolved in terrestrial microorganisms that colonise solid substrates and therefore secrete cellulases to enable penetration and utilisation of the substrate, whereas bacteria and fungi in aquatic environments would not benefit from a cell-free cellulase system and instead produce surface-bound cellulases to support their exclusive use of breakdown products as carbon and energy sources. However, evidence is emerging that in F. succinogenes, a separate and distinct mechanism is employed (Fig. 2) [118].

Microbial mechanisms of cellulose degradation. a Aerobic cell-free cellulase system (based on [75]); typical of aerobic microorganisms including T. reesei. Cellulose is hydrolysed via the synergistic interaction of individual enzymes that are secreted from the cell. b Anaerobic ‘cellulosome’ mechanism (based on [75]); typical of anaerobic bacteria (e.g. Clostridium thermocellum) and fungi. Cellulosomes consist of the catalytic enzymes capable of cellulose hydrolysis in addition to scaffoldin molecules, which anchor the enzymes to the cellulosome, and carbohydrate binding molecules (CBM) to maintain close contact with the substrate. The close proximity between the bacterial cell wall and cellulose substrate is a major benefit, resulting in concerted enzymatic activity arising from optimal synergy between cellulases. c Proposed cellulose degradation mechanism for F. succinogenes (based on [118, 134]). Attachment to the substrate is mediated by fibro-slime proteins and type IV pilin structures attached to the outer membrane. Cellulose fibres are disrupted by carbohydrate-active enzymes and individual cellulose chains are transported through the outer membrane via an ABC transporter. Current data suggests that the degradation of cellulose chains occurs in the periplasmic space
Fibrobacters are Major Degraders of Plant Biomass in the Herbivore Gut
Cellulose is the most abundant energy source on the planet, yet vertebrate herbivores do not possess the enzymes capable of degrading cellulose and other complex plant polysaccharides [89]. Consequently, herbivorous animals have evolved symbiotic relationships with bacteria, protozoa and fungi that possess the enzymes necessary for plant polymer degradation. Previous studies have indicated that the predominant species of cellulose-degrading bacteria detected via cultivation-based approaches in the herbivore gut are F. succinogenes, Ruminococcus albus and Ruminococcus flavefaciens [38, 49], notwithstanding recent studies suggesting that other as yet uncultivated bacteria may also have a role in cellulose hydrolysis within the rumen [56]. More recently, molecular biological techniques targeting the 16S rRNA gene of cellulolytic rumen bacteria have further supported the importance of F. succinogenes, R. albus and R. flavefaciens in cellulose hydrolysis [28, 93, 106, 120]. It is possible that the enzymatic system of F. succinogenes is more effective at degrading cellulose than the mechanisms used by the other cellulolytic organisms that occupy the same environment. For example, it was found that when strains S85 and A3C were grown in pure cultures, they were able to degrade a greater amount of cellulose from intact forage than the two other predominant rumen cellulolytic bacteria, R. albus and R. flavefaciens [27]. F. succinogenes is also capable of growth rate on ball-milled cellulose equivalent to that when cellobiose is used as substrate [31].
F. succinogenes has been described as one of the major cellulolytic bacterial species present in the rumen [33], and real-time polymerase chain reaction (PCR) has been widely utilised to quantify Fibrobacter spp. in the rumen [28, 58, 84, 97, 120]. Fibrobacter spp. have been detected in the intestinal tracts of a number of herbivorous species using both molecular and culture-based approaches including the bovine rumen and cecum [11, 25, 26, 47, 48, 117], ovine rumen [93, 115], porcine cecum [125], equine cecum [22, 24, 53, 63, 68], faeces of Grevy’s zebra [63], rat cecum [77, 92], black rhinoceros faeces [63], ostrich cecum [81, 82], faeces of snub-nosed monkeys [136], yak rumen [2], wild ass faeces [63], goat rumen [67], rock hyrax faeces [63], capybara faeces [63] and antelope rumen [50]. The application of 16S rRNA gene-targeted oligonucleotide probes has provided an insight into Fibrobacter diversity and ecology in a number of gut ecosystems. Lin et al. [67] applied a suite of oligonucleotide probes for quantification of Fibrobacter spp. at genus, species and subspecies level. The application of these probes to RNA extracted from cattle and goat intestinal contents indicated a greater diversity of Fibrobacter as only ca. 50% of the total Fibrobacter genus abundance could be accounted for by the species-specific probes [67]. The relative abundances of the Fibrobacter genus in this study were 0.6–6% and 0.5–2% of the total 16S rRNA for cattle and goats, respectively. A similar study of equine-associated Fibrobacter populations also demonstrated the presence of a previously undescribed population of F. succinogenes-like species in caecal contents as the genus Fibrobacter represented 12% of the total 16S rRNA, yet none of the F. succinogenes subspecies-specific probes, or the F. intestinalis probe, hybridised with RNA derived from caecal contents [68]. Bacterial 16S rRNA gene PCR amplification, cloning and sequencing of DNA extracted from the caecal contents demonstrated the presence of novel Fibrobacter spp. affiliated with F. succinogenes, but representing novel lines of descent (Fig. 1—lineage represented by sequence accession number L35547) [68].
Cellulose Degradation by Fibrobacter spp.
Electron microscopy was used to show that F. succinogenes adheres to plant cell walls and on this material forms digestive pits [16]. F. succinogenes binds tightly to the surface of plant materials via adhesins, leading to extensive plant cell wall degradation [86–88], and when adhesion cannot occur, either in non-adherent mutants [34] or due to the presence of the phenolic aldehyde vanillin, [126], cellulose degradation does not occur. The outer membrane of F. succinogenes has been found to contain 13 cellulose binding proteins, and in a mutant strain where two of these were absent the strain was able to bind to amorphous cellulose, but not crystalline cellulose [54]. When seven of these cellulose-binding proteins were absent in another mutant strain, the strain was unable to bind to either of the two forms of cellulose and no growth was detected [54]. Proteins designated as fibro-slime domain-containing proteins present on the outer membrane of F. succinogenes S85 and type IV pili may also be involved in the adherence of F. succinogenes to crystalline cellulose [118] (Fig. 2).
It is suggested that Fibrobacter spp. utilise a novel mechanism of cellulose degradation because there are genes for endocellulases, which randomly hydrolyse the cellulose chain and disrupt the crystalline structure, but not for exocellulases or processive endocellulases, both of which release cellobiose from the ends of the cellulose chains and are crucial to the established free cellulase and cellulosomal mechanisms [133]. Furthermore, genome sequence data indicate that Cytophaga hutchinsonii may utilise a similar and novel mechanism [134] and, like F. succinogenes, also exhibits gliding motility on surfaces [48]. This is intriguing because F. succinogenes is an anaerobic rumen bacterium and C. hutchinsonii an aerobic soil bacterium, and both are phylogenetically distant. This ‘third’ mechanism of cellulose depolymerisation may involve a protein complex that is present in the outer membrane of the cell, cleaving individual cellulose chains from the bound cellulose fibres and transporting them into the periplasmic space through the outer membrane. Once in the periplasmic space, the cellulose chains would then be cleaved by endoglucanases, thus eradicating the need for exocellulases or processive endocellulases [134] (Fig. 2). This would explain the requirement for the Fibrobacter cells to be bound to the cellulose as the removal and binding of the individual cellulose chains would be a key step in the mechanism. This novel mechanism has both evolutionary and biotechnological significance and may be the explanation for the superior cellulolytic ability of Fibrobacter spp. compared to that of other rumen bacteria.
The recently sequenced genome of F. succinogenes strain S85 revealed that there are numerous proteins unique to F. succinogenes; 37% of proteins could not be attributed to a known metabolic or physiological function using clusters of orthologous groups analysis [118]. Furthermore, up to 26% of the predicted proteins in the proteome of F. succinogenes did not have a known ortholog, suggesting a high content of genus- or species-specific proteins [118]. A total of 134 genes encoded enzymes that were identified by carbohydrate-active enzyme [13] analysis, representing carbohydrate esterases, carbohydrate binding modules (CBMs), polysaccharide lyases and glycosyl hydrolases derived from 49 different families. Of these, the majority were predicted to contain signal peptides, indicating that these enzymes are not targeted within the cytoplasm [118]. F. succinogenes strain S85 is predicted to have 31 cellulase genes, of which none contain the CBMs that are typically found in cellulosomes associated with adherence to crystalline cellulose. The absence of known dockerin domains in the cellulase genes and the absence of known scaffoldin genes within the genome suggest that F. succinogenes S85 does not utilise the cellulosomal degradation mechanism [118]. Whilst F. succinogenes S85 possesses endo-hemicellulases capable of hydrolysing a variety of substrates, it lacks the genes necessary to transport and metabolise any of these carbohydrates other than cellulose and its hydrolytic products [118]. F. succinogenes S85 is specialised for utilising only cellulose as growth assays utilising cellulose, pectin, starch, glucomannan, arabinogalactan and various forms of xylan found that, although all of the polysaccharides were hydrolysed, only cellulose was metabolised [118], including cellulose II, which is highly stable [130]. Forano and colleagues have studied the carbohydrate metabolism of F. succinogenes in detail (reviewed in [32]). NMR studies demonstrated the cycling of carbohydrates, notably glycogen, by F. succinogenes, in addition to several reversible metabolic pathways that enabled both the degradation and synthesis of carbohydrates. This ability to accumulate and rapidly degrade storage compounds such as glycogen may represent a strategy for rapid adaptation of F. succinogenes to changing environmental conditions. Surprisingly, F. succinogenes was found to synthesise maltodextrins and maltodextrin-1-phosphate, possibly in association with glycogen metabolism, and it is likely that the excretion of maltodextrins may support the cross-feeding of non-cellulolytic bacteria in co-culture in addition to other planktonic F. succinogenes cells [32].
A Cellulolytic Subphylum of the Fibrobacteres in the Termite Gut
It was originally thought that members of the genus Fibrobacter were restricted to the mammalian intestinal tract, but the occurrence and distribution of members of the Fibrobacteres phylum has recently been extended to include termite intestinal contents where cellulose is again the primary carbon source for the host organisms [41, 42]. However, data to support the role of symbiotic gut bacteria in the direct hydrolysis of cellulose and xylan in the termite gut were only recently reported [123].
Hongoh and colleagues [42] utilised terminal restriction fragment length polymorphism analysis in addition to general bacterial 16S rRNA gene clone libraries derived from colonies of the wood-feeding higher termite genus Microcerotermes and the lower termite genus Reticulitermes to create molecular community profiles of the bacterial gut microflora. Of 960 sequenced 16S rRNA gene clones derived from ten termite colonies (six Microcerotermes colonies and four Reticulitermes colonies), 12 phylotypes of clone sequences affiliated with the phylum Fibrobacteres were identified, and all of these sequences were from members of the higher termite genus Microcerotermes, representing approximately 10% of the total 16S rRNA clones from this group. These cloned Fibrobacteres sequences represented a novel sub-phylum cluster within the phylum, designated as Fibrobacteres subphylum 2 [42] (Fig. 1). Further work using a Fibrobacteres subphylum 2-specific probe in FISH experiments on samples of luminal fluid from the higher termite hindgut demonstrated that Fibrobacteres were the second most dominant group of the gut microflora, representing between 10.8% and 16.0% of the total bacterial cells and around 1.3 × 107 cells per gut [41]. Interestingly, FISH analysis demonstrated that the morphology of bacteria belonging to Fibrobacteres subphylum 2 differed from that of the known rumen strains of the genus Fibrobacter in that they represented undulate forms with a tapered end and a typical cell size of 0.2–0.3 × 1.3–4.9 μm [41].
Fibrobacteres subphylum 2-specific PCR primers were used to survey for these novel termite sequences in a variety of environments beyond the termite gut, including the gut of cockroaches, lake and deep sea sediments and rice paddy soil. However, Fibrobacteres subphylum 2 was not detected in any of these environments, suggesting that this novel subphylum of the Fibrobacteres represents an autochthonous lineage of termite gut symbionts [41]. Phylogenetic analysis of 16S rRNA gene sequences derived from Fibrobacteres subphylum 2 and members of the genus Fibrobacter sensu stricto (described as Fibrobacteres subphylum 1 by Hongoh et al. [41]) demonstrated 16S rRNA gene sequence similarities of 81.3% to 84.3% between subphyla 1 and 2 against 85.3% 16S rRNA gene similarity within subphylum 2 [41], again highlighting the profound genetic diversity that circumscribes this phylum. As the two currently described species of the Fibrobacteres, F. succinogenes and F. intestinalis, are known anaerobic degraders of lignocellulosic biomass in the herbivore gut, Hongoh and colleagues [41] suggested that the detection of novel lineages of Fibrobacteres in anoxic termite guts where cellulose again represents the primary carbon source for growth implies a role for these organisms in cellulolysis.
This was later confirmed when a metagenomic and functional analysis of the microbiota of a wood-feeding higher termite demonstrated the presence of a broad diversity of bacterial genes responsible for cellulose degradation, and these were identified as belonging to the phyla Spirochaetes and Fibrobacteres [128]. Fibrobacteres were detected in 16S rRNA gene inventories from the higher termite hindgut and also represented 13% of the identifiable DNA fragments from a shotgun metagenome derived from the same sample. Many of these metagenomic sequences identified as belonging to Fibrobacteres encoded glycosyl hydrolases or carbohydrate-binding modules, and proteomic analysis confirmed that some of these genes were expressed in vivo or their cloned gene modules possessed cellulase activity in vitro, implicating them in lignocellulose degradation in this environment [128]. As molecular biological and ‘omics’ techniques continue to improve our ability to characterise such communities, it is likely that the role of fibrobacters in cellulose degradation in other anoxic environments will be definitively established.
Difficulties in the Isolation and Molecular Detection of Fibrobacter spp.
Although F. succinogenes was first characterised in 1947, fibrobacters are notoriously difficult to isolate and cultivate in the laboratory, and consequently their presence in other environments has probably been greatly underestimated [84]. Undoubtedly, low cell numbers obtained by the anaerobic culture of Fibrobacter strains from the rumen have similarly resulted in the underestimation of their contribution to the degradation of cellulose [49]. Latham et al. [60] isolated several hundreds of rumen bacteria strains, but only one of these was F. succinogenes, leading them to conclude that only a small amount of the cellulolytic activity that occurred in the rumen could be ascribed to this species. Furthermore, despite ecological and physiological evidence of the importance of fibrobacters as a major degrader of plant biomass in the herbivore gut [53], it has become apparent that the nucleic acid sequences of Fibrobacter spp. are poorly represented both in 16S rRNA gene clone libraries in a number of studies on ruminant microflora [23, 120–122, 132] and a ribosomal intergenic spacer clone library [59]. In a study by Larue and colleagues [59], community DNA prepared from colonised plant biomass in the herbivore gastrointestinal tract was subjected to both ribosomal intergenic spacer analysis and denaturing gradient gel electrophoresis (DGGE). Although Fibrobacter spp. were not detected in any of the clone libraries, genus-specific PCR-DGGE for Fibrobacter spp. confirmed their presence in all community DNA samples used to generate the libraries, with the cloned sequences showing between 91% and 98% identity to previously identified F. succinogenes sequences. Furthermore, the F. succinogenes sequences were found to have no mis-matches with the oligonucleotide primers used to produce the library, indicating an inherent bias against the PCR amplification of Fibrobacter 16S rRNA gene sequences [59]. Fibrobacter spp. are often poorly represented in metagenomic studies, with some studies on the bovine rumen unable to detect any Fibrobacteres sequences at all [10, 39], although they have been detected in a number of other mammalian metagenomes [63].
Tajima et al., [120] have offered the only hypothesis thus far to explain the poor representation of Fibrobacter sequences in general bacterial 16S rRNA gene libraries. They grew pure cultures of 12 common rumen bacteria (including F. succinogenes) and added equal quantities (30 ng) of pure culture DNA to separate quantitative PCR assays with general bacterial primers. They observed that F. succinogenes was the last organism to exceed the threshold fluorescence at cycle 15.85 compared to Streptococcus bovis DNA, which surpassed the threshold fluorescence at cycle 6.74, demonstrating a prolonged amplification lag phase when compared with the other organisms. This observation was not a consequence of rRNA operon copy number as F. succinogenes possesses at least three rRNA operons compared to one copy in S. bovis. As annealing and extension of the F. succinogenes template was not affected once the threshold cycle was surpassed, the problem appears to be with the initial DNA template and it was concluded that this is possibly an effect due to DNA-associated molecules [120]. Therefore, in view of the under-representation of fibrobacters in rumen clone libraries and the difficulties in isolating these obligately anaerobic organisms, it is possible that their apparent absence from many terrestrial and aquatic anoxic environments is erroneous, particularly in environments with high cellulosic biomass content.
Molecular Detection of Fibrobacter spp. in Non-gut Environments
Members of the genus Fibrobacter are established as major degraders of lignocellulosic biomass in the herbivore gut, and the failure to detect fibrobacters in terrestrial and aquatic environments beyond this highly specialised and restricted environment supported the notion that they were in fact obligate ‘gut’ anaerobes [91]. However, the microbial-mediated depolymerisation of lignocellulose is also a feature of many other anoxic habitats in the biosphere, such as waterlogged soils, wetlands, landfill sites and the anoxic water column and sediments of freshwater, estuarine and marine systems [61]. Cellulolytic clostridia are ubiquitous within the biosphere and have been isolated from numerous environments in which cellulose is hydrolysed under anaerobic conditions, such as soils [90, 111], estuarine sediments [78, 94] freshwater sediments [62], the bovine rumen [40], methanogenic bioreactors [108, 112], waste digesters [8], anoxic rice paddy field soils [17, 129] and landfill sites [131]. This leads to the suggestion that clostridia are the predominant degraders of cellulose in the open environment. However, a number of sequences related to the Fibrobacteres phylum have been detected in general bacterial 16S rRNA gene clone libraries derived from potentially anoxic cellulose-rich environments including soils [95, 104], peat bogs [110], mangrove sediments [66] and the Atlantic and Pacific oceans [35]. Despite this, 16S rRNA gene sequences affiliated with the genus Fibrobacter (as currently defined) have until recently evaded detection, possibly due to the associated difficulties in both the isolation and molecular detection of fibrobacters. The recent detection of novel centres of variation belonging to the genus Fibrobacter in landfill sites [84] and freshwater lake sediments [83] using a genus-specific 16S rRNA gene primer set represented the first detection of fibrobacters beyond the gut. These data indicate that fibrobacters occupy a much wider ecological range than previously acknowledged and suggest a role in cellulose hydrolysis in anaerobic environments in general.
Landfill Sites
It has been suggested that anaerobic cellulose degradation in landfill sites is predominantly due to members of the genera Clostridium and Eubacterium [124]. This was first indicated by the work of Westlake et al. [131], who isolated a number of cellulolytic bacteria from landfill sites and identified them as members of these genera. Furthermore, the advent of molecular biological techniques, and specifically the use of 16S rRNA gene PCR primers, enabled further characterisation of the landfill microbiota. General bacterial 16S rRNA gene clone libraries from anaerobic landfill leachate bioreactor samples demonstrated that of those microorganisms attached to cellulosic material and in the mixed fraction, 100% and 90%, respectively, belonged to the Firmicutes and the majority of these clones fell into clusters III and XIVa of the clostridia [12]. Furthermore, 16S rRNA gene clone libraries derived from the leachate of a closed municipal solid waste landfill [44] and from the effluent leachate of a full-scale recirculating landfill [43] also did not identify any sequences belonging to the genus Fibrobacter. However, as stated above, even in the rumen where Fibrobacter are known to predominate, 16S rRNA gene clone library analysis using general bacterial primers appears to bias against the detection of fibrobacters.
Recently, novel lineages belonging to the genus Fibrobacter (as currently defined) were detected in landfill leachate samples, providing the first evidence that Fibrobacter spp. existed outside of the gut ecosystem [84]. This study utilised genus-specific 16S rRNA gene PCR primer sets targeting all known Fibrobacter spp. to detect novel sequences from the community DNA of leachate drawn from five landfill sites. Cloned PCR products were further analysed using temporal thermal gel electrophoresis and phylogenetic analysis of 58 clone sequences revealed that only two sequences could be identified as a named Fibrobacter species, and both were F. succinogenes. The remaining sequences represented novel centres of variation within the genus Fibrobacter as currently defined, occupying four distinct clusters within the genus, all of which exclusively comprised novel landfill Fibrobacter sequences (Fig. 1). Landfill Fibrobacter lineages were represented by sequence accession numbers EF186272, EF186275, EF186285 and EF186286. Of these four clusters, one contained sequences that were identified across all of the sampled sites, two contained site-specific sequences from one of two landfill sites and the fourth predominantly consisted of sequences identified from a low-level radioactive waste site in which cellulosic material was the only source of organic carbon (Fig. 1).
In this study, reverse-transcribed community RNA from landfill leachate samples was subjected to 16S rRNA gene-targeted quantitative PCR (qPCR) assays, demonstrating that the abundance of reverse-transcribed Fibrobacter 16S rRNA in landfill samples relative to total bacterial 16S rRNA could be as much as 40%. Significantly, the abundance of Fibrobacter in one landfill sample (40%) was higher than that of ovine rumen fluid samples analysed in the same way (21% to 32%). Data from this study suggested that fibrobacters are more readily detected when environmental RNA samples were used as they were detected in a greater proportion of samples when reverse-transcribed RNA was utilised in PCR reactions compared to extracted DNA [84]. As Fibrobacter spp. are considered to be predominant bacterial degraders of cellulose in the herbivore gut, it is likely that these novel lineages play a role in the degradation of cellulose that occurs in landfill environments [84]; cellulose is the main biodegradable component of landfill, representing up to 63.4% of the total organic content [9]. Recently, we have demonstrated the predominance of Fibrobacter in a cellulolytic biofilm that colonised and degraded cotton in a landfill leachate microcosm using qPCR, whereas Fibrobacter were not detected in the biofilm of an un-degraded cotton sample (unpublished data).
Although only partial Fibrobacter 16S rRNA gene sequences were obtained from landfill samples (ca. 855 bp), phylogenetic analyses suggested that these four landfill lineages represent novel centres of variation within the genus Fibrobacter as currently defined [84]. Amann and colleagues [1] suggested that Fibrobacter may in fact represent a supra-generic taxon, and the subsequent detection of novel lineages of Fibrobacteres in the termite gut and in landfill sites certainly supports this assertion. It remains necessary however, and a significant gap to our knowledge, to determine the physiology and true phylogeny of this group of organisms via the application of ‘omic’ techniques in addition to the targeted isolation and cultivation of representatives of these new taxa.
Freshwater Lakes
Novel lineages of Fibrobacter have also been detected in freshwater lakes [83, 99]. Fibrobacter genus-specific PCR and qPCR primers targeting the 16S rRNA gene demonstrated the detection of novel members of the genus Fibrobacter in lake water, sediment and colonised cotton (cellulose) samples taken from different depths of two UK freshwater lakes [83]. This study identified two sets of sequences: those that were similar to F. succinogenes (Fig. 1; lake Fibrobacter clusters similar to F. succinogenes represented by accession numbers EU468455, GU303627, EU475370 and FJ711738) and a separate and novel cluster of Fibrobacter sequences that were similar to other sequences previously observed in clone libraries from freshwater environments (Fig. 1; novel lake Fibrobacter clusters represented by accession numbers EF520548 and FJ711714).
To determine if the detection of fibrobacters in freshwater lake sediments originated from the percolation of faecal contaminants from grazing ruminants, soil and ovine faecal samples from the adjacent fields were analysed in the same way and these did not contain any sequences related to the novel ‘aquatic’ Fibrobacter lineage, suggesting that there is no linkage between the Fibrobacter sequences in these environments (Fig. 1). Furthermore, all Fibrobacter sequences clustering within the aquatic group were detected on colonised cotton samples, many of which were obtained using reverse-transcribed RNA, and both qPCR and PCR demonstrated that fibrobacters were more readily detected in colonised cotton baits than in the surrounding water or sediment sample at equivalent depth, suggesting active colonisation of cellulosic substrates and metabolic activity [83]. In addition, Fibrobacter sequences were more readily detected in the anoxic regions of the water column and sediment, consistent with the obligate anaerobic physiology of all cultivated fibrobacters. Quantitative PCR analysis of reverse-transcribed bacterial community RNA suggested low metabolic activity of Fibrobacter spp. on the colonised cotton baits (0.005% to 0.02%) and on the sediment surface (ca. 1%), although the Fibrobacter sequences were enriched on the colonised cotton baits in comparison to the surrounding water column. The preference of these aquatic Fibrobacter spp. for colonised cotton baits and lake sediment provides further support for the suggestion that these organisms contribute to the degradation of plant and algal biomass in aquatic environments [83].
Fibrobacter Cellulases in Biotechnology
Microbial cellulases have been of industrial interest for over 60 years. Initially, a fungal attack on the clothing and tents of soldiers in Southeast Asia during World War II provided the impetus to understand the mechanism of cellulase action [102]. However, the industrial focus of cellulase enzymology has recently shifted to biofuel production in the light of the current energy crisis. Cellulose is the most abundant organic polymer both in the biosphere, as a major component of plant cell walls, and in human-generated wastes and therefore represents a valuable resource. The microbial conversion of cellulose (and similar polymers) from plant matter and municipal wastes to hydrolysis products such as ethanol and glucose is an attractive vision for nations seeking alternative fuel options [74]. In addition, cellulose conversion technologies offer disposal alternatives for municipal wastes otherwise deposited in landfill sites whilst reducing the environmental impact of greenhouse gases generated from municipal waste treatment and gasoline-fuelled transport [7]. Cellulases are increasingly being utilised in second-generation biofuel pilot plants for the optimal hydrolysis of lignocellulosic materials, maximising the yield of sugars that are available for fermentation to ethanol [119].
Cellulases have a variety of industrial applications including those in food, animal feed, paper, textile, waste management, fuel and chemical industries [79]. To date, there has been research into the application of F. succinogenes cellulolytic enzymes for use in detergent additives where cellulases are utilised to brighten and soften garments [15]. F. succinogenes has also been used to produce succinic acid [64], which is utilised in a variety of industries and chemical manufacturing processes [51]. The degradative capabilities of Fibrobacter spp. are also being utilised for waste decomposition in life support systems for long-term space missions such as the Micro-Ecological Life Support Alternative [18]. Cellulolytic enzymes of Fibrobacter spp. may also be cloned into non-cellulolytic bacteria in order to improve silage production and the pretreatment of animal feeds [116]. The display of F. succinogenes β-glucanase on the cell surface of Lactobacillus reuteri is the first example of successful cloning of Fibrobacter cellulolytic enzymes into a non-cellulolytic bacterium, which was shown to improve the capability of L. reuteri to adhere to and degrade β-glucan in barley [45].
F. succinogenes cellulolytic enzymes also have the potential to be used in the production of biogas [70] and have significant potential for the refining of lignocellulosic biomass in the generation of bioethanol [73, 103]. For these processes, cellulose from plant matter and municipal waste would be utilised, thus also providing an alternative waste disposal mechanism and so reducing the environmental impact of waste treatment sites [7]. As the current work on the cellulolytic enzymes of Fibrobacter spp. is restricted to F. succinogenes, it is possible that the novel centres of variation detected in terrestrial and aquatic environments may contain cellulolytic enzymes with extended potential for applications in a variety of industrial processes, particularly in the area of second-generation biofuel production.
Final Comments
The Fibrobacteres is a diverse and functionally important phylum of bacteria, and yet there is a paucity of information on their ecology, phylogeny and physiology. This can be ascribed to the difficulties associated with the cultivation and molecular detection of members of this phylum. However, the recent application of more targeted molecular-based techniques and ‘omics’ approaches, including the use of environmental RNA rather than DNA as the starting material, has provided some important and novel observations on the Fibrobacteres phylum. Fibrobacteres are not restricted to the herbivore gut, with novel lineages detected in other anoxic environments where cellulose degradation occurs (termite gut, landfill sites and freshwater lakes). At least one species has evolved an atypical cellulose degradation mechanism, which may explain the superior hydrolytic capabilities of fibrobacters compared to other anaerobic bacterial groups. The detection of novel lineages of Fibrobacteres in termite guts, landfill sites and freshwater lakes has significant implications for their role in carbon flow in the biosphere, and their hydrolytic enzyme systems represent potential targets for novel catalysts with industrial application, such as the refining of lignocellulosic biomass for biofuel production. Isolation and cultivation of the Fibrobacteres we now know to be present and active in a number of different environments is an obvious priority.
References
Amann RI, Lin CH, Key R, Montgomery L, Stahl DA (1992) Diversity among Fibrobacter isolates—towards a phylogenetic classification. Syst Appl Microbiol 15:23–31
An DD, Dong XZ, Dong ZY (2005) Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe 11:207–215
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2005) At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71:7724–7736
Atasoglu C, Newbold CJ, Wallace RJ (2001) Incorporation of N-15 ammonia by the cellulolytic ruminal bacteria Fibrobacter succinogenes BL2, Ruminococcus albus SY3, and Ruminococcus flavefaciens 17. Appl Environ Microbiol 67:2819–2822
Bayer EA, Belaich JP, Shoham Y, Lamed R (2004) The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol 58:521–554
Bayer EA, Chanzy H, Lamed R, Shoham Y (1998) Cellulose, cellulases and cellulosomes. Curr Opin Struc Biol 8:548–557
Bayer EA, Lamed R, Himmel ME (2007) The potential of cellulases and cellulosomes for cellulosic waste management. Curr Opin Biotech 18:237–245
Benoit L, Cailliez C, Petitdemange E, Gitton J (1992) Isolation of cellulolytic mesophilic clostridia from a municipal solid-waste digester. Microbial Ecol 23:117–125
Bookter TJ, Ham RK (1982) Stabilization of solid-waste in landfills. J Env Eng Div-Asce 108:1089–1100
Brulc JM, Antonopoulos DA, Miller MEB, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Wacklin P, Coutinho PM, Henrissat B, Nelson KE, White BA (2009) Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. P Natl Acad Sci USA 106:1948–1953
Bryant MP, Robinson IM, Chu H (1959) Observations on the nutrition of Bacteroides succinogenes—a ruminal cellulolytic bacterium. J Dairy Sci 42:1831–1847
Burrell PC, O'Sullivan C, Song H, Clarke WP, Blackall LL (2004) Identification, detection, and spatial resolution of Clostridium populations responsible for cellulose degradation in a methanogenic landfill leachate bioreactor. Appl Environ Microbiol 70:2414–2419
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active enZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:233–238
Cato EP, Salmon CW (1976) Transfer of Bacteroides clostridiiformis subsp clostridiiformis (Burri and Ankersmit) Holdeman and Moore and Bacteroides clostridiiformis subsp girans (Prevot) Holdeman and Moore to genus Clostridium as Clostridium clostridiiforme (Burri and Ankersmit) comb nov—emendation of description and designation of neotype strain. Int J Syst Bacteriol 26:205–211
Chen BY, Wang HT (2008) Utility of enzymes from Fibrobacter succinogenes and Prevotella ruminicola as detergent additives. J Ind Microbiol Biot 35:923–930
Cheng KJ, Stewart CS, Dinsdale D, Costerton JW (1984) Electron-microscopy of bacteria involved in the digestion of plant-cell walls. Anim Feed Sci Tech 10:93–120
Chin KJ, Rainey FA, Janssen PH, Conrad R (1998) Methanogenic degradation of polysaccharides and the characterization of polysaccharolytic clostridia from anoxic rice field soil. Syst Appl Microbiol 21:185–200
Christophe G, Guiavarch E, Creuly C, Dussap CG (2009) Growth monitoring of Fibrobacter succinogenes by pressure measurement. Bioprocess Biosyst Eng 32:123–128
Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM (2007) The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res 35:169–172
Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, Tiedje JM (2003) The ribosomal database project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31:442–443
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:141–145
Daly K, Shirazi-Beechey SP (2003) Design and evaluation of group-specific oligonucleotide probes for quantitative analysis of intestinal ecosystems: their application to assessment of equine colonic microflora. FEMS Microbiol Ecol 44:243–252
Daly K, Stewart CS, Flint HJ, Shirazi-Beechey SP (2001) Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiol Ecol 38:141–151
Davies ME (1964) Cellulolytic bacteria isolated from large intestine of horse. J Appl Bacteriol 27:373–378
Dehority BA (1963) Isolation and characterization of several cellulolytic bacteria from in vitro rumen fermentations. J Dairy Sci 46:217–222
Dehority BA (1969) Pectin-fermenting bacteria isolated from bovine rumen. J Bacteriol 99:189–196
Dehority BA (1993) Forage cell wall structure and digestibility. Jung HG, Buxton D R, Hatfield RD, Ralph J (ed) American Society of Agronomy, Crop Science Society of America, Soil Science Society of America, Wisconsin, pp 425–453
Denman SE, McSweeney CS (2006) Development of a real-time PCR assay for monitoring anaerobic fungal and cellulolytic bacterial populations within the rumen. FEMS Microbiol Ecol 58:572–582
DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34:394–399
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Fields MW, Mallik S, Russell JB (2000) Fibrobacter succinogenes S85 ferments ball-milled cellulose as fast as cellobiose until cellulose surface area is limiting. Appl Microbiol Biot 54:570–574
Forano E, Delort AM, Matulova M (2008) Carbohydrate metabolism in Fibrobacter succinogenes: what NMR tells us. Microb Ecol Health D 20:94–102
Forsberg CW, Cheng KJ, White BA (1997) Polysaccharide degradation in the rumen and large intestine. In: Mackie RI, White BA, Isaacson RE (eds) Gastrointestinal microbiology, vol 1. Chapman & Hall, New York, pp 319–379
Gong JH, Forsberg CW (1989) Factors affecting adhesion of Fibrobacter succinogenes subsp succinogenes S85 and adherence-defective mutants to cellulose. Appl Environ Microbiol 55:3039–3044
Gordon DA, Giovannoni SJ (1996) Detection of stratified microbial populations related to Chlorobium and Fibrobacter species in the Atlantic and Pacific Oceans. Appl Environ Microbiol 62:1171–1177
Groleau D, Forsberg CW (1983) Partial characterization of the extracellular carboxymethylcellulase activity produced by the rumen bacterium Bacteroides succinogenes. Can J Microbiol 29:504–517
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Halliwell G, Bryant MP (1963) Cellulolytic activity of pure strains of bacteria from rumen of cattle. J Gen Microbiol 32:441–448
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G, Luo SJ, Clark DS, Chen F, Zhang T, Mackie RI, Pennacchio LA, Tringe SG, Visel A, Woyke T, Wang Z, Rubin EM (2011) Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331:463–467
Hobson PN, Wallace RJ (1982) Microbial ecology and activities in the rumen. Crc Cr Rev Microbiol 9:165–225
Hongoh Y, Deevong P, Hattori S, Inoue T, Noda S, Noparatnaraporn N, Kudo T, Ohkuma M (2006) Phylogenetic diversity, localization, and cell morphologies of members of the candidate phylum TG3 and a subphylum in the phylum Fibrobacteres, recently discovered bacterial groups dominant in termite guts. Appl Environ Microbiol 72:6780–6788
Hongoh Y, Deevong P, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Vongkaluang C, Noparatnaraporn N, Kudol T (2005) Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl Environ Microbiol 71:6590–6599
Huang LN, Zhou H, Zhu S, Qu LH (2004) Phylogenetic diversity of bacteria in the leachate of a full-scale recirculating landfill. FEMS Microbiol Ecol 50:175–183
Huang LN, Zhu S, Zhou H, Qu LH (2005) Molecular phylogenetic diversity of bacteria associated with the leachate of a closed municipal solid waste landfill. FEMS Microbiol Lett 242:297–303
Huang SJ, Chen MJ, Yueh PY, Yu B, Zhao X, Liu JR (2011) Display of Fibrobacter succinogenes beta-glucanase on the cell surface of Lactobacillus reuteri. J Agr Food Chem 59:1744–1751
Huang Y, Niu BF, Gao Y, Fu LM, Li WZ (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682
Hungate RE (1947) Studies on cellulose fermentation: III. The culture and isolation of cellulose-decomposing bacteria from the rumen of cattle. J Bacteriol 53:631–645
Hungate RE (1950) The anaerobic mesophilic cellulolytic bacteria. Bacteriol Rev 14:1–49
Hungate RE (1966) The rumen and its microbes. Academic, New York
Hungate RE, Phillips GD, McGregor A, Hungate DP, Buechner HK (1959) Microbial fermentation in certain mammals. Science 130:1192–1194
Isar J, Agarwal L, Saran S, Saxena RK (2006) A statistical method for enhancing the production of succinic acid from Escherichia coli under anaerobic conditions. Bioresource Technol 97:1443–1448
Jenkinson DS, Adams DE, Wild A (1991) Model estimates of CO2 emissions from soil in response to global warming. Nature 351:304–306
Julliand V, de Vaux A, Millet L, Fonty G (1999) Identification of Ruminococcus flavefaciens as the predominant cellulolytic bacterial species of the equine cecum. Appl Environ Microbiol 65:3738–3741
Jun HS, Qi M, Gong J, Egbosimba EE, Forsberg CW (2007) Outer membrane proteins of Fibrobacter succinogenes with potential roles in adhesion to cellulose and in cellulose digestion. J Bacteriol 189:6806–6815
Kobayashi Y, Shinkai T, Koike S (2008) Ecological and physiological characterization shows that Fibrobacter succinogenes is important in rumen fiber digestion—review. Folia Microbiol 53:195–200
Koike S, Kobayashi Y (2009) Fibrolytic rumen bacteria: their ecology and functions. Asian Austral J Anim 22:131–138
Koike S, Pan J, Suzuki T, Takano T, Oshima C, Kobayashi Y, Tanaka K (2004) Ruminal distribution of the cellulolytic bacterium Fibrobacter succinogenes in relation to its phylogenetic grouping. Anim Sci J 75:417–422
Koike S, Yabuki H, Kobayashi Y (2007) Validation and application of real-time polymerase chain reaction assays for representative rumen bacteria. Anim Sci J 78:135–141
Larue R, Yu ZT, Parisi VA, Egan AR, Morrison M (2005) Novel microbial diversity adherent to plant biomass in the herbivore gastrointestinal tract, as revealed by ribosomal intergenic spacer analysis and rrs gene sequencing. Environ Microbiol 7:530–543
Latham MJ, Sharpe ME, Sutton JD (1971) Microbial flora of rumen of cows fed hay and high cereal rations and its relationship to rumen fermentation. J Appl Bacteriol 34:425–434
Leschine SB (1995) Cellulose degradation in anaerobic environments. Annu Rev Microbiol 49:399–426
Leschine SB, Canaleparola E (1983) Mesophilic cellulolytic clostridia from fresh-water environments. Appl Environ Microbiol 46:728–737
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI (2008) Evolution of mammals and their gut microbes. Science 320:1647–1651
Li QA, Siles JA, Thompson IP (2010) Succinic acid production from orange peel and wheat straw by batch fermentations of Fibrobacter succinogenes S85. Appl Microbiol Biot 88:671–678
Li WZ, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Liang JB, Chen YQ, Lan CY, Tam NFY, Zan QJ, Huang LN (2007) Recovery of novel bacterial diversity from mangrove sediment. Mar Biol 150:739–747
Lin CZ, Flesher B, Capman WC, Amann RI, Stahl DA (1994) Taxon specific hybridization probes for fiber-digesting bacteria suggest novel gut-associated Fibrobacter. Syst Appl Microbiol 17:418–424
Lin CZ, Stahl DA (1995) Taxon-specific probes for the cellulolytic genus Fibrobacter reveal abundant and novel equine-associated populations. Appl Environ Microbiol 61:1348–1351
Ling JR, Armstead IP (1995) The in-vitro uptake and metabolism of peptides and amino-acids by 5 species of rumen bacteria. J Appl Bacteriol 78:116–124
Lissens G, Verstraete W, Albrecht T, Brunner G, Creuly C, Seon J, Dussap G, Lasseur C (2004) Advanced anaerobic bioconversion of lignocellulosic waste for bioregenerative life support following thermal water treatment and biodegradation by Fibrobacter succinogenes. Biodegradation 15:173–183
Ludwig W, Schleifer KH (2001) In: Boone DR, Castenholz RW (eds) Bergey’s manual of systematic bacteriology. Springer, Berlin, pp 49–65
Ludwig W, Strunk O, Klugbauer S, Klugbauer N, Weizenegger M, Neumaier J, Bachleitner M, Schleifer KH (1998) Bacterial phylogeny based on comparative sequence analysis. Electrophoresis 19:554–568
Lynd LR, Cushman JH, Nichols RJ, Wyman CE (1991) Fuel ethanol from cellulosic biomass. Science 251:1318–1323
Lynd LR, van Zyl WH, McBride JE, Laser M (2005) Consolidated bioprocessing of cellulosic biomass: an update. Curr Opin Biotech 16:577–583
Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev 66:506–577
Lynd LR, Wyman CE, Gerngross TU (1999) Biocommodity engineering. Biotechnol Progr 15:777–793
Macy JM, Farrand JR, Montgomery L (1982) Cellulolytic and non-cellulolytic bacteria in rat gastrointestinal tracts. Appl Environ Microbiol 44:1428–1434
Madden RH, Bryder MJ, Poole NJ (1982) Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium papyrosolvens sp-nov. Int J Syst Bacteriol 32:87–91
Mandels M (1985) Applications of cellulases. Biochem Soc T 13:414–416
Martinez AT, Ruiz-Duenas FJ, Martinez MJ, del Rio JC, Gutierrez A (2009) Enzymatic delignification of plant cell wall: from nature to mill. Curr Opin Biotech 20:348–357
Matsui H, Ban-Tokuda T, Wakita M (2010) Detection of fiber-digesting bacteria in the ceca of ostrich using specific primer sets. Curr Microbiol 60:112–116
Matsui H, Kato Y, Chikaraishi T, Moritani M, Ban-Tokuda T, Wakita M (2010) Microbial diversity in ostrich ceca as revealed by 16S ribosomal RNA gene clone library and detection of novel Fibrobacter species. Anaerobe 16:83–93
McDonald JE, de Menezes AB, Allison HE, McCarthy AJ (2009) Molecular biological detection and quantification of novel Fibrobacter populations in freshwater lakes. Appl Environ Microbiol 75:5148–5152
McDonald JE, Lockhart RJ, Cox MJ, Allison HE, McCarthy AJ (2008) Detection of novel Fibrobacter populations in landfill sites and determination of their relative abundance via quantitative PCR. Environ Microbiol 10:1310–1319
Miron J, Benghedalia D (1993) Digestion of cell-wall monosaccharides of ryegrass and alfalfa hays by the ruminal bacteria Fibrobacter succinogenes and Butyrivibrio fibrisolvens. Can J Microbiol 39:780–786
Miron J, Benghedalia D (1993) Digestion of structural polysaccharides of panicum and vetch hays by the rumen bacterial strains Fibrobacter succinogenes Bl2 and Butyrivibrio fibrisolvens D1. Appl Microbiol Biot 39:756–759
Miron J, Benghedalia D (1993) Untreated and delignified cotton stalks as model substrates for degradation and utilization of cell-wall monosaccharide components by defined ruminal cellulolytic bacteria. Bioresource Technol 43:241–247
Miron J, Yokoyama MT, Lamed R (1989) Bacterial-cell surface-structures involved in lucerne cell-wall degradation by pure cultures of cellulolytic rumen bacteria. Appl Microbiol Biot 32:218–222
Moir RJ (1965) The comparative physiology of ruminant-like animals. In: Dougherty RW (ed) Physiology of digestion in the ruminant. Butterworth, Washington DC, pp 1–14
Monserrate E, Leschine SB, Canale-Parola E (2001) Clostridium hungatei sp nov., a mesophilic, N-2-fixing cellulolytic bacterium isolated from soil. Int J Syst Evol Micr 51:123–132
Montgomery L, Flesher B, Stahl D (1988) Transfer of Bacteroides succinogenes (Hungate) to Fibrobacter gen-nov as Fibrobacter succinogenes comb nov and description of Fibrobacter intestinalis sp-nov. Int J Syst Bacteriol 38:430–435
Montgomery L, Macy JM (1982) Characterization of rat cecum cellulolytic bacteria. Appl Environ Microbiol 44:1435–1443
Mosoni P, Chaucheyras-Durand F, Bera-Maillet C, Forano E (2007) Quantification by real-time PCR of cellulolytic bacteria in the rumen of sheep after supplementation of a forage diet with readily fermentable carbohydrates: effect of a yeast additive. J Appl Microbiol 103:2676–2685
Murray WD, Hofmann L, Campbell NL, Madden RH (1986) Clostridium lentocellum sp-nov, a cellulolytic species from river sediment containing paper-mill waste. Syst Appl Microbiol 8:181–184
Nusslein K, Tiedje JM (1999) Soil bacterial community shift correlated with change from forest to pasture vegetation in a tropical soil. Appl Environ Microbiol 65:3622–3626
O’Sullivan AC (1997) Cellulose: the structure slowly unravels. Cellulose 4:173–207
Ozutsumi Y, Tajima K, Takenaka A, Itabashi H (2006) Real-time PCR detection of the effects of protozoa on rumen bacteria in cattle. Curr Microbiol 52:158–162
Paster BJ, Ludwig W, Weisburg WG, Stackebrandt E, Hespell RB, Hahn CM, Reichenbach H, Stetter KO, Woese CR (1985) A phylogenetic grouping of the Bacteroides, Cytophagas, and certain Flavobacteria. Syst Appl Microbiol 6:34–42
Percent SF, Frischer ME, Vescio PA, Duffy EB, Milano V, McLellan M, Stevens BM, Boylen CW, Nierzwicki-Bauer SA (2008) Bacterial community structure of acid-impacted lakes: what controls diversity? Appl Environ Microbiol 74:1856–1868
Qi M, Nelson KE, Daugherty SC, Nelson WC, Hance IR, Morrison M, Forsberg CW (2005) Novel molecular features of the fibrolytic intestinal bacterium Fibrobacter intestinalis not shared with Fibrobacter succinogenes as determined by suppressive subtractive hybridization. J Bacteriol 187:3739–3751
Qi M, Nelson KE, Daugherty SC, Nelson WC, Hance IR, Morrison M, Forsberg CW (2008) Genomic differences between Fibrobacter succinogenes S85 and Fibrobacter intestinalis DR7, identified by suppression subtractive hybridization. Appl Environ Microbiol 74:987–993
Reese ET, Siu RGH, Levinson HS (1950) The biological degradation of soluble cellulose derivatives and its relationship to the mechanism of cellulose hydrolysis. J Bacteriol 59:485–497
Rubin EM (2008) Genomics of cellulosic biofuels. Nature 454:841–845
Saul DJ, Aislabie JM, Brown CE, Harris L, Foght JM (2005) Hydrocarbon contamination changes the bacterial diversity of soil from around Scott Base, Antarctica. FEMS Microbiol Ecol 53:141–155
Shah HN, Collins MD (1983) Genus Bacteroides—a chemotaxonomical perspective. J Appl Bacteriol 55:403–416
Shinkai T, Kobayashi Y (2007) Localization of ruminal cellulolytic bacteria on plant fibrous materials as determined by fluorescence in situ hybridization and real-time PCR. Appl Environ Microbiol 73:1646–1652
Shinkai T, Ohji R, Matsumoto N, Kobayashi Y (2009) Fibrolytic capabilities of ruminal bacterium Fibrobacter succinogenes in relation to its phylogenetic grouping. FEMS Microbiol Lett 294:183–190
Shiratori H, Reno H, Ayame S, Kataoka N, Miya A, Hosono K, Beppu T, Ueda K (2006) Isolation and characterization of a new Clostridium sp that performs effective cellulosic waste digestion in a thermophilic methanogenic bioreactor. Appl Environ Microbiol 72:3702–3709
Sipat A, Taylor KA, Lo RYC, Forsberg CW, Krell PJ (1987) Molecular-cloning of a xylanase gene from Bacteroides succinogenes and its expression in Escherichia coli. Appl Environ Microbiol 53:477–481
Sizova MV, Panikov NS, Tourova TP, Flanagan PW (2003) Isolation and characterization of oligotrophic acido-tolerant methanogenic consortia from a Sphagnum peat bog. FEMS Microbiol Ecol 45:301–315
Skinner FA (1960) The isolation of anaerobic cellulose-decomposing bacteria from soil. J Gen Microbiol 22:539–554
Sleat R, Mah RA, Robinson R (1984) Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans sp-nov. Appl Environ Microbiol 48:88–93
Stahl DA, Flesher B, Mansfield HR, Montgomery L (1988) Use of phylogenetically based hybridization probes for studies of ruminal microbial ecology. Appl Environ Microbiol 54:1079–1084
Stewart CS, Bryant MP (1988) The rumen microbial ecosystem. Hobson PN (ed) Elsevier Appl Sci, New York, pp 21–75
Stewart CS, Duncan SH (1985) The effect of avoparcin on cellulolytic bacteria of the ovine rumen. J Gen Microbiol 131:427–435
Stewart CS, Flint HJ (1989) Bacteroides (Fibrobacter) succinogenes, a cellulolytic anaerobic bacterium from the gastrointestinal-tract. Appl Microbiol Biotech 30:433–439
Stewart CS, Paniagua C, Dinsdale D, Cheng KJ, Garrow SH (1981) Selective isolation and characteristics of Bacteriodes succinogenes from the rumen of a cow. Appl Environ Microbiol 41:504–510
Suen G, Weimer PJ, Stevenson DM, Aylward FO, Boyum J, Deneke J, Drinkwater C, Ivanova NN, Mikhailova N, Chertkov O, Goodwin LA, Currie CR, Mead D, Brumm PJ (2011) The complete genome sequence of Fibrobacter succinogenes S85 reveals a cellulolytic and metabolic specialist. Plos One 6:e18814. doi:10.1371/journal.pone.0018814
Sun Y, Cheng JY (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Bioresource Technol 83:1–11
Tajima K, Aminov RI, Nagamine T, Matsui H, Nakamura M, Benno Y (2001) Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl Environ Microbiol 67:2766–2774
Tajima K, Aminov RI, Nagamine T, Ogata K, Nakamura M, Matsui H, Benno Y (1999) Rumen bacterial diversity as determined by sequence analysis of 16S rDNA libraries. FEMS Microbiol Ecol 29:159–169
Tajima K, Arai S, Ogata K, Nagamine T, Matsui H, Nakamura M, Aminov RI, Benno Y (2000) Rumen bacterial community transition during adaptation to high-grain diet. Anaerobe 6:273–284
Tokuda G, Watanabe H (2007) Hidden cellulases in termites: revision of an old hypothesis. Biol Lett 3:336–339
Van Dyke MI, McCarthy AJ (2002) Molecular biological detection and characterization of Clostridium populations in municipal landfill sites. Appl Environ Microbiol 68:2049–2053
Varel VH, Fryda SJ, Robinson IM (1984) Cellulolytic bacteria from pig large-intestine. Appl Environ Microbiol 47:219–221
Varel VH, Jung HJG (1986) Influence of forage phenolics on ruminal fibrolytic bacteria and in vitro fiber degradation. Appl Environ Microbiol 52:275–280
Vogels GD (1979) Global cycle of methane. A Van Leeuw J Microb 45:347–352
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang XN, Hernandez M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450:560–565
Weber S, Lueders T, Friedrich MW, Conrad R (2001) Methanogenic populations involved in the degradation of rice straw in anoxic paddy soil. FEMS Microbiol Ecol 38:11–20
Weimer PJ, French AD, Calamari TA (1991) Differential fermentation of cellulose allomorphs by ruminal cellulolytic bacteria. Appl Environ Microbiol 57:3101–3106
Westlake K, Archer DB, Boone DR (1995) Diversity of cellulolytic bacteria in landfill. J Appl Bacteriol 79:73–78
Whitford MF, Forster RJ, Beard CE, Gong JH, Teather RM (1998) Phylogenetic analysis of rumen bacteria by comparative sequence analysis of cloned 16S rRNA genes. Anaerobe 4:153–163
Wilson DB (2008) Three microbial strategies for plant cell wall degradation. In: Wiegel J, Maier R, Adams M (eds) Incredible anaerobes: From physiology to genomics to fuels. Wiley-Blackwell, Oxford, pp 289–297
Wilson DB (2009) Evidence for a novel mechanism of microbial cellulose degradation. Cellulose 16:723–727
Woese CR, Stackebrandt E, Macke TJ, Fox GE (1985) A phylogenetic definition of the major eubacterial taxa. Syst Appl Microbiol 6:143–151
Wu CF, Yang F, Gao RC, Huang ZX, Xu B, Dong YY, Hong T, Tang XH (2010) Study of fecal bacterial diversity in Yunnan snub-nosed monkey (Rhinopithecus bieti) using phylogenetic analysis of cloned 16S rRNA gene sequences. Af J Biotechnol 9:6278–6289
Yilmaz S, Haroon MF, Rabkin BA, Tyson GW, Hugenholtz P (2010) Fixation-free fluorescence in situ hybridization for targeted enrichment of microbial populations. ISME J 4:1352–1356
Acknowledgements
Research on fibrobacters by the authors has been funded by the Natural Environment Research Council (AJM and JEM) and the Systematics Association’s SynTax award scheme, supported by the Linnean Society of London, BBSRC and NERC (JEM). ERJ is supported by a 125th Anniversary Scholarship at Bangor University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ransom-Jones, E., Jones, D.L., McCarthy, A.J. et al. The Fibrobacteres: an Important Phylum of Cellulose-Degrading Bacteria. Microb Ecol 63, 267–281 (2012). https://doi.org/10.1007/s00248-011-9998-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-011-9998-1