
Abstract
Oral microbiota plays a vital role in maintaining the homeostasis of oral cavity. Dental caries are among the most common oral diseases in children and pathogenic bacteria contribute to the development of the disease. However, the overall structure of bacterial communities in the oral cavity from children with dental caries has not been explored deeply heretofore. We used high-throughput barcoded pyrosequencing and PCR-denaturing gradient gel electrophoresis (DGGE) to examine bacterial diversity of oral microbiota in saliva and supragingival plaques from 60 children aged 3 to 6 years old with and without dental caries from China. The multiplex barcoded pyrosequencing was performed in a single run, with multiple samples tagged uniquely by multiplex identifiers. As PCR-DGGE analysis is a conventional molecular ecological approach, this analysis was also performed on the same samples and the results of both approaches were compared. A total of 186,787 high-quality sequences were obtained for evaluating bacterial diversity and 41,905 unique sequences represented all phylotypes. We found that the oral microbiota in children was far more diverse than previous studies reported, and more than 200 genera belonging to ten phyla were found in the oral cavity. The phylotypes in saliva and supragingival plaques were significantly different and could be divided into two distinct clusters (p < 0.05). The bacterial diversity in oral microbiome analyzed by PCR-DGGE and barcoded pyrosequencing was employed to cross validate the data sets. The genera of Streptococcus, Veillonella, Actinomyces, Granulicatella, Leptotrichia, and Thiomonas in plaques were significantly associated with dental caries (p < 0.05). The results showed that there was no one specific pathogen but rather pathogenic populations in plaque that significantly correlated with dental caries. The enormous diversity of oral microbiota allowed for a better understanding of oral microecosystem, and these pathogenic populations in plaque provide new insights into the etiology of dental caries and suggest new targets for interventions of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent advances in metagenomics have shown that microorganisms associated with the human body are approximately ten times more numerous than our own cells and contain, in aggregate, about 100 times more genes [1]. This has led to the suggestion that humans and our microbial symbionts should be considered “supraorganisms” [1, 2]. Some microbes cause disease, but the overwhelming majority are either innocuous or play a vital role in human physiological processes, including the immune response, digestion and vitamin production [3–8]. As one of the largest and most complex human-associated microbial habitats, the oral cavity harbors large numbers of bacteria that can have important effects on health. During the past 40 years, a wealth of knowledge has been gathered about these bacteria: over 250 oral species have been isolated and characterized by cultivation, and over 450 species have been identified by culture-independent molecular approaches [9, 10]. Within the human body, the interplay between oral microbiota and the host is associated with oral health [11] as well as contributes to several systemic diseases, such as bacterial endocarditis [12], pneumonia [13], preterm low birth weight [14, 15], and coronary heart disease [16, 17]. Data from these studies suggest that the microbiota in the oral cavity may be crucial to the health or disease status of the human host.
Dental caries are one of the most common childhood diseases [18, 19]. They form over time because of an interaction between acid-producing bacteria and fermentable carbohydrate. Additional host factors in the teeth and saliva contribute to carie development. The disease develops in both crowns and roots of teeth, and it can arise in early childhood as an aggressive form that affects the primary teeth of infants and toddlers. Risk for caries includes physical, biological, environmental, behavioral, and lifestyle-related factors such as high numbers of cariogenic bacteria, inadequate salivary flow, insufficient fluoride exposure, poor oral hygiene, inappropriate methods of feeding infants, and poverty. Dental plaque, a complex structurally and functionally organized microbial biofilm, is required for the formation of dental caries.
Three major hypotheses have been developed to explain the etiology of dental caries: the specific plaque hypothesis, the nonspecific plaque hypothesis, and the ecological plaque hypothesis [20–22]. Several studies have shown that dental caries might be caused by potentially pathogenic microbial communities rather than a single pathogen [23, 24]. With our inability to cultivate most of the microbial species residing in the oral cavity, however, the taxonomic composition, its community structure, and, ultimately, its function have not been fully understood. Thus, understanding the etiology and pathogenesis of the disease is challenging. With the advent of molecular techniques, bacterial diversity and community structure in different microhabitats have been investigated using molecular fingerprinting methods such as PCR-DGGE and sequence analysis of microbial 16S rRNA genes and other universal targets (such as cpn60) [25–29]. The normal bacterial microbiota in the oral cavity has been defined by cloning and sequencing approaches. The bacteria there are highly diverse and oral cavity site and individual subject specific [9]. Becker et al. identified several species or phylotypes that might play a role in early childhood caries by sequence analysis of PCR-amplified bacterial 16S rDNA [28]. However, routinely used cloning and sequencing techniques are time consuming and labor intensive and only detect predominant members of the communities studied. More recently, the bacterial diversity in the healthy adult oral cavity was examined with high-throughput barcoded parallel 454 pyrosequencing. The data showed that the bacterial phylotypes were more complex than previously reported [30]. Improvements in pyrosequencing enable a dramatic increase in throughput via parallel in-depth analysis of large scale samples with limited sample processing and lower costs [31]. However, the overall extent of bacterial diversity in different oral diseases, such as dental caries, periodontal diseases, gingivitis, dentoalveolar abscesses, etc., has not yet been extensively studied. Therefore, further study of bacterial diversity in oral cavity, especially microbiota in saliva and plaque with advanced technology, may provide insights for understanding the development of dental caries.
To estimate the detailed bacterial diversity of oral community of the children with and without dental caries, and to identify the key population changes relevant to dental caries development, we first utilized PCR-DGGE fingerprinting with broad-range primers corresponding to the bacterial 16S rRNA hypervariable V3 region to investigate the bacterial communities in children with dental caries. We then applied massively parallel pyrosequencing, combined with a DNA barcoding system, to characterize the oral microbiome in saliva and supragingival plaque samples from 60 children in China. Our experiment will help us define the overall structure of oral microbiota in children with dental caries and find out the potential pathogenic populations in dental caries and provide new insights into the disease.
Methods
Subject Selection
Sixty children from two kindergartens in Hangzhou City, Zhejiang province, China (34 males and 26 females) were recruited for this study in November 2008. Their ages ranged from 3 to 6 years old. All subjects were examined clinically before sampling and were subsequently divided into four groups: MN group (males without caries, n = 17), MC group (males with caries, n = 17), FN group (females without caries, n = 11), and FC group (females with caries, n = 15). Written informed consent was obtained from the parents or guardians of all participants prior to enrollment, with approval of the ethical committee of the First Affiliated Hospital, College of Medicine, Zhejiang University. The definition and diagnosis of dental caries were based on the criteria of the World Health Organization (WHO); the WHO TRS-621 C-version periodontal probe was used to confirm visual evidence of caries. Subjects from MN and FN groups had no caries or existing restorations. A carious lesion was recorded present when a lesion had an unmistakable cavity, undermined enamel, decayed softened floor or wall, or felt soft or leathery on probing. A dental restoration with secondary decay was also recorded as a caries lesion. Each tooth was given one of the following scores: sound (score 0), decayed (score 1), filled with decay (score 2) (details in SI Table: Clinical Data). All participants were instructed not to clean their teeth the evening and morning before sampling. Any subject who met the following criteria was excluded from participation: less than 18 teeth present, use of antibiotics in the previous 3 months, or active bacterial or viral infections in other parts of the body.
Sample Collection and Preparation
Sampling was performed in the morning before the participants ate breakfast. Participants were asked to expectorate into a sterile cryogenic vial (Corning, NY, USA). A 1-ml spontaneous, unstimulated whole saliva specimen was obtained by each individual. Skilled dentists collected supragingival plaque samples by scraping the dental surfaces with a sterile metal loop approximately 20 times from the carious teeth. The metal loop was cut off and immersed in 1 ml of saline solution that had been UV-irradiated to avoid DNA contamination. Theses samples were placed on liquid nitrogen immediately and stored at −80°C until use.
Total Bacterial Genomic DNA Extraction
Bacteria were pelleted from saliva and dental plaque samples by centrifugation (Thermo Electron Corporation, Boston, MA, USA) at full speed (more than 10,000×g) for 10 min. The pellets were re-suspended in lysis buffer and homogenized with 100 mg of zirconium beads (0.1 mm) in Mini-beadbeater (FastPrep, Thermo Electron Corporation, USA) for 2 min. Bacterial DNA was extracted using QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions with minor modification (details in SI Methods). All DNA was stored at −20°C before further analysis.
PCR-DGGE Analysis
The V3 regions of 16S rRNA genes were amplified with universal bacterial primers (341F 5′-GTATTACCGCGGCTGCTGG-3′, 534R 5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGGACTCCTACGGGAGGCAGCAG-3′) using the hot-start touchdown protocol described by Muyzer et al. [32], and the reaction mixtures and the amplification program described previously by Li et al. [33] with minor modifications. The DGGE analysis (with 28% to 58% gradient) and the sequence analysis of the excised DGGE bands were carried out as described earlier [33]. The similarities of PCR-DGGE DNA profiles were analyzed with Quantity One® 1-D Analysis software (version 4. 6.2; Bio-Rad Laboratory, Hercules, CA, USA). A similarity matrix was constructed using Dice's similarity coefficient. Dendrograms were constructed by the unweighted pair group method, using arithmetic averages (UPGMA) (details in SI Methods).
454 Pyrosequencing and Data Analysis
PCR amplification of the 16S rDNA hypervariable V3-region was performed with universal bacterial primers 341f [34] –518r [32], which correspond to positions 341 to 534 in Escherichia coli as described earlier (It must be noted that 454 adaptor sequences and barcodes are not shown here) [35]. Amplicon pyrosequencing was performed with standard 454/Roche GS-FLX protocols [36]. After pyrosequencing, all reads were screened and filtered for quality and length using customized Perl scripts written by ourselves. Raw sequences were processed and analyzed following the procedure described previously [37]. Sequences were assigned to samples by examining the 8-bp barcode (Table S1) [38]. The qualified 16S rRNA gene fragments were processed as previously described [39]. OTUs and OTU rarefaction curves were created by aligning unique tag sequences and used to determine richness and diversity indexes (Shannon Weaver and Simpson diversity indices), ACE, Chao1, Good’s coverage at each dissimilarity level using MOTHUR (version 1.5.0) (http://schloss.micro.umass.edu/mothur/Main_Page) [40]. Read level taxonomic assignments were performed using the Ribosomal Database Project (RDP) Classifier program (http://rdp.cme.msu.edu/) [41] with an 80% bootstrap score. Community comparative analysis was performed using the web-based service UniFrac [42] and principal component analysis (PCA) was performed using SPSS Data Analysis Program version 12.0 (SPSS Inc, Chicago, IL, USA). The neighbor-joining tree was constructed using the MEGA 4.0 program based on the Jukes-Cantor model and used for UniFrac analysis. The statistical significance of differences in microbial community composition, and Shannon and Simpson index between sample categories was determined by SPSS with one-way ANOVA (details in SI Methods).
Results
PCR-DGGE Analysis of Oral Microbiota in Children with and Without Caries
PCR-DGGE was a useful tool for detecting changes of predominant microbiota in specific microhabitats and has been widely applied for comparative analysis of parallel samples. The PCR-DGGE profiles were obtained by amplifying bacterial DNA from saliva and dental plaques of 60 children with and without caries. Each lane represented one subject, which was selected from its group at random. As shown in Fig. 1a, the PCR-DGGE profiles of children with and without caries revealed significant differences in the overall structure and composition of oral microbiome. The pattern of bands revealed more richness in dental plaques than in saliva. Figure 1b depicts the results of a Ward’s analysis in which the Dice coefficient for measuring similarity algorithm in banding patterns was applied. The plaque and saliva samples displayed statistically significant clustering of profiles (clusters I and II—Fig. 1b). The PCR-DGGE profiles in males and females with caries were not significantly different. The fragments of interest (29 bands in the DGGE gel represented the predominant members of oral cavity) were excised from the DGGE lanes, reamplified, sequenced, and identified by BLAST with the 16S rRNA V3 region sequences. As the universal bacterial primers were used for total bacterial genomic DNA amplification, it should be noted that only predominant members of the bacterial community were represented in the samples.

a PCR-DGGE analysis of the predominant bacterial communities in saliva (S) and dental plaques (P) from male and female children with dental caries active and caries free. Each lane represented one subject which was selected in its group at random. Bands that were marked in the DGGE gel were identified by cloning and sequencing to facilitate the interpretation of the figure. Bands 1 uncultured Cytophaga sp., 2 uncultured Streptococcus sp., 3 unidentified oral bacterium RP55-4, 4 Aggregatibacter aphrophilus, 5 uncultured Haemophilus sp., 6 uncultured Streptococcus sp., 7 Streptococcus pneumoniae, 8 uncultured Bacteroidetes bacterium, 9 uncultured Bacteroidetes bacterium, 10 uncultured Atopobium sp., 11 uncultured Bacteroidetes bacterium, 12 uncultured Neisseria sp., 13 uncultured Veillonella sp., 14 Aggregatibacter aphrophilus, 15 uncultured Veillonella sp., 16 uncultured Veillonella sp., 17 uncultured Streptococcus sp., 18 uncultured Prevotella sp., 19 uncultured Prevotella sp., 20 uncultured Rothia sp., 21 uncultured Lautropia sp., 22 Actinomyces naeslundii, 23 uncultured Lautropia sp., 24 Actinobacterium MSB3008, 25 Actinomyces naeslundii, 26 Actinomyces naeslundii, 27 Actinomyces naeslundii, 28 uncultured Rothia sp., 29 uncultured Lautropia sp. b Dendrogram of the DGGE profiles shown in a. MN males without caries, MC males with caries, FN females without caries, FC females with caries, S saliva, P dental plaques
Based on the PCR-DGGE profiles, the predominant microbiota in each group of samples were similar, although slight inter-individual variation existed. Species identification from the bands of saliva and plaque was highly associated. In all subjects, Streptococcus spp., Veillonella spp., Prevotella spp., Lautropia spp., and Rothia spp. were the predominant microbiota in saliva and dental plaque. Actinomyces spp. was present in most of the plaque samples. Based on the DGGE analysis, however, we could not quantitatively compare the bacterial populations in these samples. There was no significant difference in the diversity of predominant microbiota in caries-free vs. caries-active children in saliva or dental plaques.
Results of the Pyrosequencing Data
In our study, we obtained 186,787 pyrosequencing tags that passed our quality control methods, and these were used for data analysis. Specifically, we got 72,831 sequences from dental plaque samples and 113,956 sequences from saliva samples. The average length of the sequences was 145 bp after trimming the primers. The average number of sequence reads was 1,557 per sample. Total unique sequences from the eight groups numbered 41,905 and represented all phylotypes. It was unexpected that we obtained more sequences from saliva samples than from plaque samples because the same amount of bacterial DNA was mixed from each sample before preparing the single-stranded DNA library for pyrosequencing. It can be assumed that the biased ratio of the eight groups was not only because of an inefficiency in the emPCR technique for different species of oral microbiota but also because of the impurities or unknown compounds disturbing the accurate measurement of DNA amount [35, 43].
The Diversity of Oral Microbiota in Caries-Active and Caries-Free Children
The numbers of species detected in a sample, or the numbers of organisms discerned at any given phylogenetic level, are strongly affected by the number of sequences analyzed. The richness of the total bacterial communities in saliva and plaque were estimated by rarefaction curves. Our present data showed slight higher bacterial diversity in saliva groups than in dental plaque groups, but the shape of the rarefaction curves (Fig. 2) indicated that bacterial richness of the saliva and plaque samples were not yet completely revealed by this number of sequences; additional sampling will be required to determine the true microbial diversity of the oral cavity. We found that there were about 2,000 phylotypes by clustering at 3% dissimilarity level and about 1,000 phylotypes from each group by clustering at 5% dissimilarity level; there were more phylotypes in saliva than in dental plaque samples from the same groups. For the eight groups of oral communities analyzed at 3% dissimilarity level, the number of OTU detected was close to the total number of OTU estimated by Chao1 and ACE diversity indices, additional evidence that the natural communities were well covered by the sequencing effort (Table 1). Good’s coverage was around 95% for the all sequences in all eight groups, indicating that about five additional phylotypes would be expected for every 100 additional sequenced reads. This level of coverage indicated that the 16S rRNA sequences identified in these groups represent the majority of bacterial sequences present in the samples of saliva and dental plaque in the current study. With the unique sequences analyzed, saliva samples and plaque samples in each group had similar overall levels of diversity respectively (p > 0.05 for eight groups, parametric ANOVA for Shannon and Simpson index) (Fig. 3), but the composition of the bacterial communities was significantly different between saliva and plaque (Fig. 4, Fig. S1 and Table 1). The samples in each group or individual sample was divided into two clusters based on UniFrac metrics. We found that samples in saliva groups formed a cluster very distinct from the plaque groups, indicating that they harbored different microbial communities. Comparisons of the cluster analysis with the phylogenetic tree and the PCA between two groups showed similar results for the distances based on genus composition (Fig. S2).

a Rarefaction curves were used to estimate richness (i.e., number 0.03 dissimilarity bacterial taxa) among the eight groups. The vertical axis shows the number of OTUs that would be expected to be found after sampling the number of tags or sequences shown on the horizontal axis. b The x-axis indicates the individual OTUs (at 0.03 dissimilarity level), ranked according to their relative abundance (high to low). The y-axis indicates the cumulative abundance of the OTUs

Shannon Weaver index and Simpson index were used to estimate diversity (i.e., a combined assessment of the number of 1% dissimilarity bacterial taxa and their abundance) among the eight groups. Data shown as mean with SEM. There were no statistically significant differences between the eight groups by parametric ANOVA

Differentiation in oral bacterial communities from eight groups of caries-active and caries-free children. Community differentiation was measured by using the unweighted UniFrac algorithm; the scale bar indicates the distance between clusters in UniFrac units. All of the branch nodes shown here were found to be significant (p < 0.001), indicating that saliva and dental plaque harbored distinct bacterial communities
Composition and Distribution of Oral Microbiota Observed with 454 Pyrosequencing
The phylogenetic classification of sequences from saliva and dental plaque by phylum is summarized in Fig. 5. Ten phyla were found in oral microbiota in children with or without caries. The vast majority of sequences belonged to one of the eight major phyla: Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Fusobacteria, Spirochaetes, or candidate division TM7 and SR1. Of the eight major phyla, Firmicutes (around 23–42% of total sequences in each group) and Bacteroidetes (around 16–37% of total sequences in each group) were two of the predominant microbiota in oral cavity, and were overrepresented in saliva (except for Firmicutes in FN-P group). Proteobacteria, Actinobacteria, and Fusobacteria were more abundant in dental plaque. These five phyla had different abundances in saliva and dental plaque (p < 0.05). No one phylum among the five above was significantly different between caries-active and caries-free samples (p > 0.05). The remaining bacteria belonged to the phyla Spirochaetes and candidate division TM7 or SR1 (around 0.3–0.4% of total sequences). The two rare phyla of Tenericutes and Cyanobacteria were only found in saliva samples with low relative abundance in the total sequences.

The relative abundance of oral bacterial V3 tags obtained by pyrosequencing in saliva and dental plaque from children with caries active and caries free by phylum. Phylogenetic classification for the pyrosequencing analysis obtained from a Ribosomal Database Project Classifier analyses
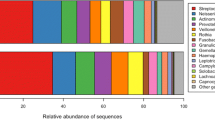
At the genus level, sequences from saliva and plaque represented 203 different genera, while 153 different genera were found in dental plaque (120 genera in caries-active samples and 116 genera in caries-free samples) and 156 different genera were found in saliva (132 genera in caries-active samples and 115 genera in caries-free samples), respectively (Fig. 6). Among these genera, 13 genera (Actinomyces, Capnocytophaga, Corynebacterium, Fusobacterium, Haemophilus, Granulicatella, Kingella, Leptotrichia, Neisseria, Prevotella, Streptococcus, Thiomonas, and Veillonella) constituted roughly 80% of the oral microbiota. Of all genera obtained from oral cavity, Atopobium, Actinomyces, Corynebacterium, Capnocytophaga, Prevotella, Fusobacterium, Leptotrichia, Campylobacter, Erysipelothrix, Oribacterium, Peptostreptococcus, Rothia, and Streptococcus were significantly different between saliva and dental plaque (p < 0.05). In our study, six genera (Streptococcus, Veillonella, Actinomyces, Granulicatella, Leptotrichia, and Thiomonas), which constituted a large proportion of oral microbiota, were significantly different between caries-active and caries-free samples in plaque (p < 0.05) (Fig. 7). This observation suggests that dental plaque bacterial communities including these genera are involved in caries development in children.

Heatmap analyses of the 72 predominant bacterial genera detected using 16S rRNA gene-based pyrosequencing among eight groups of caries-active and caries-free saliva and dental plaque samples in male and female children. The abundance plot shows the proportion of 16S rRNA gene pyrosequences in each of the samples

Genus detected from saliva and supragingival plaque in children with caries active and caries free. Among these predominant genera in oral cavity, Streptococcus, Veillonella, Actinomyces, Granulicatella, Leptotrichia, and Thiomonas in supragingival plaque were associated with dental caries significantly (p < 0.05). The salivary microbiota maintained a stable level, and no one genus significantly correlated with dental caries (p > 0.05)
At the species level, defined as OTUs at 3% dissimilarity level, around 1,600 to 2,800 phylotypes were found in saliva and plaque, respectively. While a more conservative approach, defined as OTUs at 5% dissimilarity level, about 1,000 phylotypes were found. The eight groups shared a great degree of community similarity (approximately 20%) in oral cavity at 3% dissimilarity level (Fig. 8).

One example of Venn diagrams for overlap between FC-P (plaque from female children with caries) observed OTUs vs. FC-S (saliva from female children with caries) observed OTUs. The Venn diagrams show the overlap in all OTUs calculated at the 0.03 dissimilarity level. The number of OTUs in group FC-P is 1,630. The number of OTUs in group FC-S is 2,593. The number of OTUs shared between groups FC-P and FC-S is 812. Percentage of OTUs that are shared in groups FC-P and FC-S is 23.81%
Comparison of Pyrosequencing with PCR-DGGE Analysis
The bacterial diversity in oral cavity of children was analyzed by PCR-DGGE fingerprinting and barcoded pyrosequencing. These phylotypes detected by both methods in the samples of saliva and plaque correlated well with each other with two molecular analytical methods. PCR-DGGE fingerprinting, which is a conventional molecular ecological approach, only detected the predominant microbiota (Streptococcus, Prevotella, Neisseria, Haemophilus, Thiomonas, Veillonella, Atopobium, Rothia, Actinomyces, and Lautropia) in the oral cavity. These phylotypes were also the most abundant among the 454 pyrosequencing reads. However, the pyrosequencing analysis revealed more diverse bacterial communities, as there were 41,905 phylotypes revealed with the pyrosequencing analysis but not with PCR-DGGE fingerprinting. Pyrosequencing provided a high-throughput, high-depth approach to analyze the 16S rRNA gene sequences and explore bacterial diversity in different microhabitats, which can be used in detecting minor populations in oral microbiota. As only predominant microbiota could be detected by PCR-DGGE, band richness could not reveal the overall extent of bacterial diversity in the oral cavity. With clustering analysis using UniFrac algorithm, we found similar cluster profiles in the eight groups (Fig. 4). The samples in the saliva and plaque were divided into two clusters, respectively (Fig. 1b).
Discussion
Comprehensive investigation of the composition of the oral microbial ecosystem is essential for understanding the etiology and achieving the prevention and treatment of dental caries. Our study represented for the first time the extensive examination of bacterial diversity in oral cavity and dental caries in children under age of 6. Although there are several previous studies that focus on the oral microbiota in dental caries of children, molecular analysis such as the broad-range PCR assays, reverse-capture checkerboard hybridization, and PCR-clone library analysis still cannot demonstrate the overall structure and composition of oral microbiota comprehensively [28, 44–46]. PCR-DGGE is one of the molecular fingerprinting methods targeting of the hypervariable regions of 16S rDNA directly that were capable of surveying entire bacterial communities without cultivation [32]. With this technique, we found that Streptococcus, Prevotella, Neisseria, Haemophilus, Thiomonas, Veillonella, Atopobium, Rothia, Actinomyces, and Lautropia constituted the predominant microbiota in saliva and plaque of children. Among these genera, potential cariogenic genera [46] such as Streptococcus, Actinomyces, and Veillonella constituted the larger proportion of oral microbiota. There was a small increase in the number of bands detected by DGGE in plaque compared with saliva, particularly in the high G + C% region (Fig. 1a). Previous studies have shown a significantly greater diversity of oral microbes in caries-free individuals compared with caries-active individuals [47]. However, our data showed no similar gel pattern in the DGGE profiles and bacterial communities from oral cavity were demonstrated to be much more complex than previously [47]. These differences might be caused by different primer sets selected targeting the different hypervariable regions of 16S rRNA genes. The hypervariable region(s) of chosen for amplification can greatly influence the PCR-DGGE profiles and diversity indices produced from community DNA samples, and even subtle differences in primer sequences can result in substantially different profiles and assessment of microbial diversity [48]. By comparing of different hypervariable regions of 16S rRNA genes for PCR-DGGE, Yu and Morrison have shown that the DGGE profiles of the V3 region were best [48]. From our findings, the absence, or comparative faintness of bands in the high G + C% region of saliva profiles, suggests that salivary microbiota, which includes the major plaque genus Actinomyces, was present in proportionately lower levels than in plaque. And, there was a limit for detecting bands in the DGGE profiles of complex communities [49]. Some minor bacterial populations in samples might not be detected by this method and not all the species have been detected in a single individual’s oral cavity. From the complex DGGE profiles obtained in our study, we could not find any one band that was specifically associated with caries.
In order to the make up for the limitations of PCR-DGGE, we carried out barcoded parallel high-throughput 454 pyrosequencing for the extensive study of oral microbiota in children. In fact, the depth of the bacterial diversity in a specific microhabitat was strongly influenced by the total number of sequences that used for analysis [50]. These previous studies obtained only hundreds of reads for each sample and severely limited the depth of understanding of the overall structure of oral microbiota. However, pyrosequencing of 16S rRNA gene amplicons for microbial community profiling can, for equivalent costs, yield more than two orders of magnitude more sensitivity than traditional PCR cloning and Sanger sequencing [51]. And, in our study, multiplex barcoded pyrosequencing analysis enables us to analyze an increased number of samples at a time, to obtain more reads in a single run and to perform in-depth analyses for studies of comparative microbial ecology. Although the pyrosequencing read lengths of the sequences were significantly short compared with the sequences obtained with traditional Sanger sequencing methods (600–800 bp), these short sequences (about 145 bp in our study) provided not only excellent coverage but also excellent recovery for classification at the genus level [52]. In addition, the 145 bp in highly variable region V3 of the 16S rRNA gene sequences had good discerning power (provided that a 1 bp difference in the 16S rRNA gene sequences differentiated the reads by 0.55% for bacteria) and were long enough to be sufficient for assigning the taxa [35]. Engelbrektson et al. have shown that for a given number of reads, shorter 16S rRNA gene amplicons yield greater species richness than do longer amplicons. Approximately 100-bp amplicons produced significantly higher estimates of richness than 400 or 1,000-bp products did [53]. All indicated that multiplex parallel 454 pyrosequencing offered a highly automated, rapid, economical, and accurate method for the analysis of bacterial diversity, and our study represented for one of the most extensive investigations of bacterial diversity in the oral cavity.
With barcoded parallel pyrosequencing, we achieved a view of the oral bacterial diversity from children at a much deeper level. However, despite the depth of our surveys, our diversity estimates still represented only the lower estimates of phylotypes richness in oral cavity. We did not reach the saturation level in the rarefaction curves, indicating that the true diversity was greater than that we identified (Fig. 2). Our diversity analysis is restrained by the depth of our sequencing. Recently, a computational model revealed that more than 9 million unique genes are likely to be present in the human gut bacterial community. This is far greater richness than had been expected [54]. The most common phylotypes accounted for more than 95% of sequences and less than 5% of sequences represented the other phylotypes identified in the oral microhabitat, indicative of low abundance [30, 55]. To date, only 700 species have been reported in the oral cavity using both culture-dependent and culture-independent approaches [9, 10]. These previous study are severely underestimating the actual diversity in the subjects. Good’s estimated coverage showed that most of phylotypes (95%) were identified in saliva and plaque of caries-active and caries-free subjects. These phylotypes could represent the majority of bacterial sequences present in the oral cavity of children and were used to explore the complex etiology of dental caries deeply. The oral microbiota that colonizes the oral cavity changes with age and developmental status such as primary and permanent tooth growth [56, 57]. Compared with the oral microbiota of healthy adults determined similarly by cultivation-independent 16S rDNA sequence analysis, our oral cavity results from children show lower diversity [30]. Although the total numbers of sequences obtained in our study (186,787 reads) were somewhat fewer than that obtained from oral cavity of healthy adults (197,673 reads), we obtained more sequences per sample (1,557 vs. 1,170) and more unique sequences (strain level) in children (41,905 vs. 28,978). In this sense, the oral microbiota from saliva and plaque of children in our study were more representative than that from the previous study [30]. Zaura et al. have showed that a major proportion of oral bacterial sequences of three unrelated healthy individuals by 454 pyrosequencing was identical and indicated that there was “core microbiome” in the oral microbiome [27]. More subjects (60 children) participated in our study and significant inter-individual variations were found. No core microbiome was found that correlated with dental caries of children.
Using the RDP Classifier, the total sequences were assigned into ten phyla and 203 different genera. We found that the oral microbiota in children could be divided into saliva and plaque clusters, and several phylotypes were significantly different between saliva and plaque. All these children lived in similar environment circumstances and shared their kindergarten environment. No gender differences in oral microbiome diversity were detected. In our study, the taxonomic composition of oral microbiota in children constituted five predominant phyla. These five phyla showed significant differences between saliva and plaque, but the proportion of these phyla in saliva and plaque was not in accord with the previous study in healthy adults [30]. Intriguingly, Proteobacteria (genus Neisseria and Thiomonas in β-Proteobacteria; Haemophilus in γ-Proteobacteria), which were more abundant in saliva in healthy adults, were higher in plaque in children. The transition of these bacterial profiles could be a consequence of the age related oral cavity changes. Our data suggest that the Streptococcus, Granulicatella and Veillonella, Actinomyces, Leptotrichia, and Thiomonas genera constituted a large proportion of oral microbiota and may play an important role in the development of dental caries. Streptococcus, which was one of the predominant microbiota in oral cavity, showed the property of coaggregation with other bacteria such as Veillonella [58–60]. Previous studies have shown that Streptococcus consisted of a large numbers of species, including Streptococcus oralis, Streptococcus mitis, Streptococcus sobrinus, and Streptococcus mutans. Among these species in Streptococcus, non-S. mutans such as S. oralis and S. mitis accounted for a large proportion, while cariogenic bacteria such as S. mutans and S. sobrinus only accounted for a small proportion of the bacteria in plaque from caries-free individuals (0.04% and 0.00%) and caries-active individuals (0.9% and 0.03%) [61]. Although Streptococci were less abundant in plaque from subjects with caries, the cariogenic bacterial species of Streptococcus and other putative cariogenic bacteria such as Veillonella, Actinomyces, and Leptotrichia increased significantly and accounted for a large proportion of the bacteria in the oral microbiota of such subjects. Veillonella, utilizing lactic acid produced by Streptococcus as a carbon source, were increased in plaque from subjects with caries along with the cariogenic bacterial species of Streptococcus [58, 62] but have a limited ability to adhere to host tissue. A number of observations have shown that the diversity and richness of Veillonella affected the oral health, not only for dental caries, but also for periodontal disease [63]. As predicted previously, there was a high level of correlation between the numbers of Veillonella and the numbers of Streptococcus. Some species of Veillonella such as Veillonella alcalescens have shown synergistic effect with S. mutans in aggravating the process of dental caries [59]. Another genus in Firmicutes, the Granulicatella, one of the most fastidious oral bacteria, and a possible cause of infective endocarditis [64], was decreased significantly in plaque from individuals with caries. In the oral microbiota, Granulicatella might contribute to the initial adherence of enamel surfaces [65], and the proportion change might be associated with dental caries. Surprisingly, the genus of Lactobacillus (phylum Firmicutes), already identified as cariogenic bacteria, was almost absent in our study. As the genera Streptococcus and Lactobacillus were acid-producing cariogenic bacteria, these two genera were abundant in advanced caries in the previous studies [44, 66]. Lactobacillus, anaerobic bacteria, might be abundant in deep-dentin lesions with lower redox potential but not in supragingival plaque and saliva. Our results were also consistent with those from a previous study of healthy adults [30]. Previous studies have shown that the genus of Actinomyces, in accompaniment with Streptococcus, participated in the initial stages of dental plaque formation and played a major role in the process of dental carie formation [46, 67]. Anaerobic Actinomyces bacteria also preferentially colonized the deeper region of the plaque [67]. Actinomyces are heterofermenters but tend to become homolactic producers under anaerobic conditions, thus contributing to enamel demineralization and further tooth decay. Leptotrichia coaggregated with the potential cariogenic bacteria such as Veillonella and Streptococcus and were significantly associated with dental caries [46]. We also found that the genus of Thiomonas in plaque was associated with caries. Previous studies have shown that Propionigenium, a putative etiological agent of root caries in elderly patients [68], was not significantly associated with dental caries in primary teeth of children [46]. Other predominant bacteria in the oral cavity, such as Rothia, Porphyromonas, Prevotella, Capnocytophaga, and Neisseria, might be involved in the susceptibility of an individual to periodontal disease [63, 69, 70]. With these bacteria mentioned above, the potential cariogenic bacteria interacted with each other in oral cavity, accelerated the formation of dental plaque, and participated in the process of dental caries. It was clearly observed in our study that there was no specific pathogen but pathogenic populations in supragingival plaque that significantly associated with dental caries. These pathogenic populations interacted with each other in plaque. The resulting microecological equilibrium varied from eubiosis to dysbiosis and led to the development of dental caries. Our results supported the ecological plaque hypothesis in the process of dental carie development.
In our present study, we did not detect a significant association between salivary microbiota and dental caries, although there were a large number of carie associated genera in saliva. This result was unexpected, as the cariogenic bacteria such as S. mutans and Lactobacillus have been most commonly detected from saliva and are frequently used as an indicator to evaluate the clinical activity of dental caries. Our results also confirmed the similar findings of a previous study performed in children [45]. The microbiota in saliva constituted several predominant genera such as Streptococcus, Prevotella, Neisseria, Porphyromonas, Haemophilus, Veillonia, and Rothia, which likely contributes to supragingival plaque formation but does not directly participate in the process of dental carie formation. However, the continuous flow of saliva acts directly to clear the acids produced by cariogenic bacteria eliminating enamel dissolution. Saliva additionally supplies a number of diverse salivary constituents that have “anticaries activity” such as saliva SIgA. In this sense, saliva plays a vital role in protecting teeth and antagonizing the development of dental caries. As oral microbiota in saliva and dental plaque exist in the same spatial environment, they interacted intimately and the salivary microbiota likely influences the community composition of dental plaque [23]. Although the composition of microbiota varied among individuals in our study, the salivary microbiota was not significantly different between caries-active and caries-free subjects. For some cariogenic bacteria that are obligate anaerobes, the local environment in oral cavity affects the bacterial species within the microbial community [9]. Saliva, which also contained antibacterial components, may be not suitable for the propagation of these bacteria. The composition of salivary microbiota might participate in the process of periodontal disease, but apparently not in dental caries in children [63].
In summary, the bacterial diversity of predominant oral microbiota in children was investigated by PCR-DGGE, and the overall structure of oral microbiome was comprehensively explored by high-throughput parallel barcoded 454 pyrosequencing. We found that the oral microbiota was far more diverse than previous studies, and more than 200 genera belonging to ten phyla were found in the oral cavity. Our results showed that the oral microbiota was significantly different between saliva and plaque in children (p < 0.05), independent of the presence or absence of caries. The cariogenic communities in dental plaque including the genus of Streptococcus, Veillonella, Actinomyces, Granulicatella, Leptotrichia, and Thiomonas were significantly associated with dental caries and might play important roles in the process of carie development. The enormous microbial richness of oral microbiota in children provided novel insights into the potential pathogenic communities in the development of dental caries and may help design more effective interventions for the disease.
References
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE (2006) Metagenomic analysis of the human distal gut microbiome. Science 312:1355–1359
Friedrich MJ (2008) Microbiome project seeks to understand human body's microscopic residents. JAMA 300:777–778
Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449:811–818
Cash HL, Whitham CV, Behrendt CL, Hooper LV (2006) Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313:1126–1130
Ley RE, Peterson DA, Gordon JI (2006) Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124:837–848
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL (2005) An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122:107–118
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE (2005) Defining the normal bacterial flora of the oral cavity. J Clin Microbiol 43:5721–5732
Paster BJ, Olsen I, Aas JA, Dewhirst FE (2006) The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000 42:80–87
Ruby J, Goldner M (2007) Nature of symbiosis in oral disease. J Dent Res 86:8–11
Lockhart PB, Durack DT (1999) Oral microflora as a cause of endocarditis and other distant site infections. Infect Dis Clin North Am 13:833–850, vi
Paju S, Scannapieco FA (2007) Oral biofilms, periodontitis, and pulmonary infections. Oral Dis 13:508–512
Boggess KA, Beck JD, Murtha AP, Moss K, Offenbacher S (2006) Maternal periodontal disease in early pregnancy and risk for a small-for-gestational-age infant. Am J Obstet Gynecol 194:1316–1322
Offenbacher S, Boggess KA, Murtha AP, Jared HL, Lieff S, McKaig RG, Mauriello SM, Moss KL, Beck JD (2006) Progressive periodontal disease and risk of very preterm delivery. Obstet Gynecol 107:29–36
Beck JD, Eke P, Heiss G, Madianos P, Couper D, Lin D, Moss K, Elter J, Offenbacher S (2005) Periodontal disease and coronary heart disease: a reappraisal of the exposure. Circulation 112:19–24
Beck JD, Eke P, Lin D, Madianos P, Couper D, Moss K, Elter J, Heiss G, Offenbacher S (2005) Associations between IgG antibody to oral organisms and carotid intima-medial thickness in community-dwelling adults. Atherosclerosis 183:342–348
Anusavice KJ (2002) Dental caries: risk assessment and treatment solutions for an elderly population. Compend Contin Educ Dent 23:12–20
Selwitz RH, Ismail AI, Pitts NB (2007) Dental caries. Lancet 369:51–59
Loesche WJ (1992) The specific plaque hypothesis and the antimicrobial treatment of periodontal disease. Dent Update 19(68):70–72
Marsh PD (1994) Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res 8:263–271
Theilade E (1986) The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol 13:905–911
Jenkinson HF, Lamont RJ (2005) Oral microbial communities in sickness and in health. Trends Microbiol 13:589–595
Kuramitsu HK, He X, Lux R, Anderson MH, Shi W (2007) Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev 71:653–670
Hill JE, Goh SH, Money DM, Doyle M, Li A, Crosby WL, Links M, Leung A, Chan D, Hemmingsen SM (2005) Characterization of vaginal microflora of healthy, nonpregnant women by chaperonin-60 sequence-based methods. Am J Obstet Gynecol 193:682–692
Schellenberg J, Links MG, Hill JE, Dumonceaux TJ, Peters GA, Tyler S, Ball TB, Severini A, Plummer FA (2009) Pyrosequencing of the chaperonin-60 universal target as a tool for determining microbial community composition. Appl Environ Microbiol 75:2889–2898
Zaura E, Keijser BJ, Huse SM, Crielaard W (2009) Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol 9:259
Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, Boches SK, Dewhirst FE, Griffen AL (2002) Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol 40:1001–1009
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, Baker CC, Di Francesco V, Howcroft TK, Karp RW, Lunsford RD, Wellington CR, Belachew T, Wright M, Giblin C, David H, Mills M, Salomon R, Mullins C, Akolkar B, Begg L, Davis C, Grandison L, Humble M, Khalsa J, Little AR, Peavy H, Pontzer C, Portnoy M, Sayre MH, Starke-Reed P, Zakhari S, Read J, Watson B, Guyer M (2009) The NIH human microbiome project. Genome Res 19:2317–2323
Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, ten Cate JM, Crielaard W (2008) Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res 87:1016–1020
Meyer M, Stenzel U, Hofreiter M (2008) Parallel tagged sequencing on the 454 platform. Nat Protoc 3:267–278
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, Wei H, Chen Y, Lu H, Zuo J, Su M, Qiu Y, Jia W, Xiao C, Smith LM, Yang S, Holmes E, Tang H, Zhao G, Nicholson JK, Li L, Zhao L (2008) Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 105:2117–2122
Watanabe K, Kodama Y, Harayama S (2001) Design and evaluation of PCR primers to amplify bacterial 16S ribosomal DNA fragments used for community fingerprinting. J Microbiol Methods 44:253–262
Roh SW, Kim KH, Nam YD, Chang HW, Park EJ, Bae JW (2010) Investigation of archaeal and bacterial diversity in fermented seafood using barcoded pyrosequencing. ISME J 4:1–16
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R (2008) Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods 5:235–237
Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M, Fire AZ (2007) A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res 35:e130
Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci USA 103:12115–12120
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Lozupone C, Hamady M, Knight R (2006) UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform 7:371
Binladen J, Gilbert MT, Bollback JP, Panitz F, Bendixen C, Nielsen R, Willerslev E (2007) The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS ONE 2:e197
Corby PM, Lyons-Weiler J, Bretz WA, Hart TC, Aas JA, Boumenna T, Goss J, Corby AL, Junior HM, Weyant RJ, Paster BJ (2005) Microbial risk indicators of early childhood caries. J Clin Microbiol 43:5753–5759
Li Y, Ge Y, Saxena D, Caufield PW (2007) Genetic profiling of the oral microbiota associated with severe early-childhood caries. J Clin Microbiol 45:81–87
Aas JA, Griffen AL, Dardis SR, Lee AM, Olsen I, Dewhirst FE, Leys EJ, Paster BJ (2008) Bacteria of dental caries in primary and permanent teeth in children and young adults. J Clin Microbiol 46:1407–1417
Li Y, Ku CY, Xu J, Saxena D, Caufield PW (2005) Survey of oral microbial diversity using PCR-based denaturing gradient gel electrophoresis. J Dent Res 84:559–564
Yu Z, Morrison M (2004) Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl Environ Microbiol 70:4800–4806
Ercolini D (2004) PCR-DGGE fingerprinting: novel strategies for detection of microbes in food. J Microbiol Methods 56:297–314
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, Kent AD, Daroub SH, Camargo FA, Farmerie WG, Triplett EW (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1:283–290
Tringe SG, Hugenholtz P (2008) A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11:442–446
Liu Z, DeSantis TZ, Andersen GL, Knight R (2008) Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 36:e120
Engelbrektson A, Kunin V, Wrighton KC, Zvenigorodsky N, Chen F, Ochman H, Hugenholtz P (2010) Experimental factors affecting PCR-based estimates of microbial species richness and evenness. ISME J (in press)
Yang X, Xie L, Li Y, Wei C (2009) More than 9,000,000 unique genes in human gut bacterial community: estimating gene numbers inside a human body. PLoS ONE 4:e6074
Andersson AF, Lindberg M, Jakobsson H, Backhed F, Nyren P, Engstrand L (2008) Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 3:e2836
Papaioannou W, Gizani S, Haffajee AD, Quirynen M, Mamai-Homata E, Papagiannoulis L (2009) The microbiota on different oral surfaces in healthy children. Oral Microbiol Immunol 24:183–189
Tanner AC, Milgrom PM, Kent R Jr, Mokeem SA, Page RC, Riedy CA, Weinstein P, Bruss J (2002) The microbiota of young children from tooth and tongue samples. J Dent Res 81:53–57
Chalmers NI, Palmer RJ Jr, Cisar JO, Kolenbrander PE (2008) Characterization of a Streptococcus sp.–Veillonella sp. community micromanipulated from dental plaque. J Bacteriol 190:8145–8154
Egland PG, Palmer RJ Jr, Kolenbrander PE (2004) Interspecies communication in Streptococcus gordonii–Veillonella atypica biofilms: signaling in flow conditions requires juxtaposition. Proc Natl Acad Sci USA 101:16917–16922
Al-Ahmad A, Follo M, Selzer AC, Hellwig E, Hannig M, Hannig C (2009) Bacterial colonisation of enamel in situ investigated with fluorescence in situ hybridization (FISH). J Med Microbiol (in press)
Choi EJ, Lee SH, Kim YJ (2009) Quantitative real-time polymerase chain reaction for Streptococcus mutans and Streptococcus sobrinus in dental plaque samples and its association with early childhood caries. Int J Paediatr Dent 19:141–147
Arif N, Sheehy EC, Do T, Beighton D (2008) Diversity of Veillonella spp. from sound and carious sites in children. J Dent Res 87:278–282
Takeshita T, Nakano Y, Kumagai T, Yasui M, Kamio N, Shibata Y, Shiota S, Yamashita Y (2009) The ecological proportion of indigenous bacterial populations in saliva is correlated with oral health status. ISME J 3:65–78
Ohara-Nemoto Y, Kishi K, Satho M, Tajika S, Sasaki M, Namioka A, Kimura S (2005) Infective endocarditis caused by Granulicatella elegans originating in the oral cavity. J Clin Microbiol 43:1405–1407
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR (2007) Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA 104:13780–13785
Chhour KL, Nadkarni MA, Byun R, Martin FE, Jacques NA, Hunter N (2005) Molecular analysis of microbial diversity in advanced caries. J Clin Microbiol 43:843–849
Dige I, Raarup MK, Nyengaard JR, Kilian M, Nyvad B (2009) Actinomyces naeslundii in initial dental biofilm formation. Microbiology 155:2116–2126
Preza D, Olsen I, Aas JA, Willumsen T, Grinde B, Paster BJ (2008) Bacterial profiles of root caries in elderly patients. J Clin Microbiol 46:2015–2021
Ready D, D'Aiuto F, Spratt DA, Suvan J, Tonetti MS, Wilson M (2008) Disease severity associated with presence in subgingival plaque of Porphyromonas gingivalis, Aggregatibacter actinomycetemcomitans, and Tannerella forsythia, singly or in combination, as detected by nested multiplex PCR. J Clin Microbiol 46:3380–3383
Yilmaz O (2008) The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology 154:2897–2903
Acknowledgements
This present work was funded by the grant of National Basic Research Program of China (973 Program) No. 2007CB513001, and partly supported by grants from China’s National Science and Technology Major Project (No. 2008ZX10002-009, No. 2008ZX10004-002 and 2009ZX10004-105). We thank Dr. Michael Brownstein and Dr. Liliana Losada for critical reading and useful suggestions.
Author information
Authors and Affiliations
Corresponding authors
{kind=link}
{kind=link}
Rights and permissions
About this article
Cite this article
Ling, Z., Kong, J., Jia, P. et al. Analysis of Oral Microbiota in Children with Dental Caries by PCR-DGGE and Barcoded Pyrosequencing. Microb Ecol 60, 677–690 (2010). https://doi.org/10.1007/s00248-010-9712-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-010-9712-8