
Abstract
A method was developed for enriching bacterial cells from soybean stems which was recalcitrant for a culture-independent analysis of bacterial community due to the interference with plant DNA. Stem homogenates were fractionated by a series of differential centrifugations followed by a Nycodenz density gradient centrifugation. The efficiency of bacterial cell enrichment was assessed by ribosomal intergenic spacer analysis (RISA). The intensity and the number of bacterial amplicons of RISA were markedly increased in the DNA extracted from the enriched bacterial cells compared to that in the DNA directly extracted from soybean stems. The phylogenetic diversity of the enriched bacterial cells was evaluated by analyzing a clone library of 16S rRNA gene in comparison with those of the culturable fractions of the enriched and non-enriched stem-associated bacteria, endophytic bacteria, and epiphytic bacteria. The results indicated that the method was able to enrich both endophytic and epiphytic bacteria from soybean stems, and was useful to assess the bacterial diversity based on a 16S rRNA gene clone library. When the sequence data from all clones (1,332 sequences) were combined, 72 operational taxonomic units were affiliated with Proteobacteria (Alpha-, Beta-, and Gammaproteobacteria), Actinobacteria, Firmicutes, and Bacteroidetes, which also provided the most comprehensive set of data on the bacterial diversity in the aerial parts of soybeans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The phytosphere is an attractive habitat for microorganisms because of the availability of many nutrients and its environmental stability. Although useful information have been provided by culture-dependent methodologies for the microbial diversity in these plant-associated environments, the application of culture-independent methodologies has now revealed that the majority of plant-associated microbes have not yet been cultured in the laboratory [3, 26, 60]. With soybean-associated microbial communities, characterization to date has been based mainly on culture-dependent methods [18, 28, 39, 45]. Recently, we found that the nodulation phenotype of soybeans had a marked impact on the microbial diversity in the rhizosphere [20]. However, our analysis of stem-associated microbes failed because of serious interference by plant DNA in the PCR amplification, despite of the importance of shoot-associated bacteria, which can affect the growth, and development of plants [31, 32].
Plant DNA and PCR inhibitors present in a plant tissue are main causes of interferences for culture-independent analyses of microbes in the phytosphere [45]. Studies of the aerial parts of plants like leaves, stems, and seeds have been particularly hampered by this technical difficulty, because these tissues contain less amount of microbial biomass and the chloroplast DNA often outcompetes with bacterial 16S rRNA gene as a template DNA in PCR amplification [11, 34, 51, 52].
In order to reduce the contamination of plant DNA and PCR inhibitor substances in a sample, there have been several attempts to extract microbial cells from the surface or inside of plant tissues [15, 24, 34, 42, 60]. One of the methods is extracting bacteria by releasing cells from the surface of plant tissues by simply washing and/or ultrasonic treatment in the case of the community analysis of the phyllosphere [24, 60], or from the apoplastic sap of a pieces of stems in the case of the endosphere [15, 42]. Although these studies successfully analyzed the bacterial community in the phyllosphere or endosphere in culture-independent methods, it is not clear that how much extent of the bacterial diversities could be covered by these methods relative to the whole bacterial community in the aerial parts of plants.
An alternative procedure to overcome the interference of plant DNA is to use a primer set specific to bacteria. Chelius and Tripllet [8] developed a primer (799f) designed for the specific amplification of bacterial 16S rRNA gene sequences directly from root DNA, and this primer has been successfully used in previous studies [19, 40]. However, Rasche et al. [41] pointed out that their 16S rRNA gene libraries from sweet pepper, generated by using the 799f primer, contained large numbers of clones assigned as chloroplast sequences. Becker et al. [5] also reported that mitochondrial 16S rRNA gene could be amplified by PCR with 799f primer in potato. These results indicate that the specificity of the primer is also dependent on the genotypes of plant organelles. We found that this was also the case for soybeans. Thus, the majority of amplicons with 16S rRNA gene primers were exclusively derived from chloroplast sequences, although there were sequence differences between the primer 799f and the 16S rRNA gene of soybean chloroplast DNA. Another approach regarding a bacterial-specific primer is the use of a taxon-specific primer. The successful application of taxon-specific primers has been shown in several reports [3, 43, 52, 56]. However, this strategy is currently not available for all taxonomic groups, and is restricted to only known phylogenetic groups, excluding unknown taxons which may be detected only by using universal primers.
Jiao et al. [23] introduced an idea of enriching uncultured bacterial cells from plant tissues by enzymatic hydrolysis of the plant cell wall, followed by differential centrifugation. Subsequently, Wang et al. [58] have reported the successful enrichment of plant-associated bacteria from tree bark by a relatively simple method using a detergent and a salting-out procedure for eliminating plastid and plant debris. These results indicate good feasibility for the usefulness of a bacterial cell enrichment method for studying the diversity and metagenome analysis of plant-associated bacteria.
The aims of this work were (1) to develop a technique suitable for enriching bacterial cells from stems of field-grown soybeans and (2) to evaluate the phylogenic diversities of stem-associated bacteria by a clone library analysis of bacterial 16S rRNA gene.
Materials and Methods
Plant Materials
Soybean seeds (Glycine max ‘Enrei’) were planted on 29 May 2007 in an experimental field at Tohoku University (Kashimadai, Miyagi, Japan). The stems were harvested on 11 August 2007 and immediately transported on ice to the laboratory. The plants were serially washed well with tap and distilled water, and the leaves were removed manually. Stems were stored at −80°C until use. A flow chart of the experimental design is shown in Fig. 1.

A flow chart of the experimental design of 16S rRNA gene clone library construction. Two libraries were constructed from the bacterial cell enrichment fraction (gray libraries), and EB and CEB stand for enriched bacteria and cultured enriched bacteria, respectively. Three libraries were from the conventional culture methods (white libraries) and CDB, CEP, and CEN stand for cultured directly prepared bacteria, epiphytic bacteria, and endophytic bacteria, respectively. The cross indicates the failure of a clone library directly constructed from the DNA of soybean stems pulverized by liquid nitrogen. Cultivation was carried out by plating out and incubating the samples on nutrient agar plates for 5 days at 25°C
Preparation of Endophytic and Epiphytic Bacterial Cells
Epiphytic and endophytic bacteria were cultured from the soybean stems as follows. A 10-g piece of stem (5 cm long) was placed in a 50-ml conical flask and shaken together with 20 ml of an extraction buffer (0.85% NaCl, 0.01% Tween 20) for 1 h at 4°C. The resulting supernatant (epiphytic bacterial cell fraction) was serially diluted and plated onto nutrient agar (Difco Laboratories, Becton Dickinson Microbiology Systems, Sparks, MD, USA) without NaCl (NA). Subsequently, the same stems were immersed in 70% EtOH for 1 min and then in 5% NaOCl for 5 min. After being repeatedly washed with sterilized water, the stems were ground in liquid nitrogen with a mortar and pestle. The resulting stem powder (endophytic bacterial cell fraction) was serially diluted with nutrient broth (NB) (Difco Laboratories) and plated onto NA. After incubation at 25°C for 5 days, the bacterial cells from the three NA plates containing 100 to 200 visible colonies were collected by rinsing with 10 ml of sterile water. All treatments were carried out in triplicate, and the collected bacterial cells from plates were pooled separately as epiphytic or endophytic bacterial cell fractions. The pooled bacterial cells were mixed very well with a vortex stirrer in a 50-ml conical tube and pelleted by centrifugation with a TOMY TMA-S26 rotor (TOMY, Tokyo, Japan) at 10,000 rpm for 1 min at 10°C, and a portion of the pellet (0.1 g of fresh weight cells) was used for extracting DNA and for constructing the clone libraries of 16S rRNA genes for culturable epiphytes and endophytes (libraries CEP and CEN in Fig. 1).
Preparation from Enriched and Non-enriched Bacterial Cells
Stems (100 g) were homogenized in 500 ml of bacterial cell extraction (BCE) buffer (50 mM Tris–HCl [pH 7.5], 1% Triton X-100, 2 mM 2-mercaptoethanol [added just prior to use]) in a blender (Model 911 Clamshell; Hamilton Beach/Proctor-Silex, Southern Pines, NC, USA) on high speed for three 1-min periods and allowed to cool on ice for 1 min between each blending period. The homogenate was filtered through a layer of sterilized Miracloth (CalBiochem, La Jolla, CA, USA). The filtrate was then transferred to a clean centrifugation tube.
The filtrate was then centrifuged at 500×g (1,800 rpm with a Hitachi RPR9-2 rotor) for 5 min at 10°C. The supernatant was transferred to a clean tube without disturbing the loose pellet and was centrifuged at 5,500×g (6,000 rpm with a Hitachi RPR9-2 rotor) for 20 min at 10°C. The supernatant was discarded, and the pellet was suspended in 50 ml of BCE buffer with a vortex stirrer. After filtration of the suspension through a layer of sterilized Kimwipe (Kureshia, Tokyo, Japan) to remove unsoluble particles, the filtrate was centrifuged at 10,000×g (10,000 rpm with a Hitachi RPR20-2 rotor) for 10 min at 10°C. The supernatant was discarded and the pellet was suspended in 50 ml of BCE buffer. After the suspension had been filtered through a layer of sterilized Kimwipe, the steps of high-speed centrifugation and filtration were repeated. The final filtrate was suspended in 6 ml of 50 mM Tris–HCl (pH 7.5).
Then the suspension was overlaid on 4 ml of Nycodenz (AXIS-SHIELD PoC AS, Oslo, Norway) solution (8 g of Nycodenz dissolved in 10 ml of 50 mM Tris–HCl (pH 7.5)) and centrifuged at 10,000×g (9,000 rpm with a Hitachi RPS40T rotor) for 40 min at 10°C. After centrifugation, the whitish band located at the interface of the upper and lower phases was collected as a bacterial cell fraction. The bacterial suspension (approximately 500 µl) was mixed with an equal volume of sterilized water in a 1.5-ml microtube. The sample was then centrifuged with a TOMY TMA-S26 rotor (TOMY) at 10,000 rpm for 1 min at 10°C.
The bacterial pellet was used for extracting DNA and for constructing a clone library of 16S rRNA genes as enriched bacteria for the library EB in Fig. 1. A portion of the enriched bacterial cells was also serially diluted and plated onto NA plates. After incubation at 25°C for 5 days, the bacterial cells from the NA plates containing 100 to 200 visible colonies were collected by rinsing with 10 ml of sterile water. Then, bacterial cells collected from the NA plates were pooled. The pooled bacterial cells was pelleted by centrifugation with a TOMY TMA-S26 rotor at 10,000 rpm for 1 min at 10°C, and a portion of the pellet (0.1 g of fresh weight cells) was used for extracting DNA and for constructing clone libraries of the 16S rRNA gene for the culturable fraction of enriched bacteria (library CEB in Fig. 1). All treatments were done in triplicate.
A culturable bacterial fraction of non-enriched sample was also prepared. Unsterilized stems (50 g) were ground in liquid nitrogen with a mortar and pestle. The resulting stem powder (0.2 g) was used to extract DNA as well as to make a serial dilution for plating onto NA plates. After incubation of the plates at 25°C for 5 days, the bacterial cells were collected from plates containing 100 to 200 visible colonies and were used to construct a clone library as a culturable fraction directly derived from bacteria in the stem powder considered as a non-enriched sample (library CDB in Fig. 1).
Microscopic Observation
The enriched bacterial cells were stained with DAPI (4′,6′-diamidino-2-phenylindole). The specimens were then photographed under a fluorescence microscope (BX51; Olympus, Tokyo, Japan) equipped with fluorescence mirror units U-MNUA2 (Olympus) for those stained with DAPI.
DNA Extraction
Total bacterial DNA was prepared from each bacterial cell fraction described in the above sections by using a DNA extraction method [22]. The final DNA samples were resuspended in 100 µl of sterilized water. The quality and quantity of DNA were assessed spectrophotometrically by calculating absorbance at a wavelength of 260 nm (A 260) and A 260/A 230 and A 260/A 280 ratios.
Ribosomal Intergenic Spacer Analysis (RISA)
RISA was performed as previously described [46] by using bacterial primers ITSF/ITSReub [6]. We selected RISA amplicons that were consistent among triplicate samples for cloning and sequencing as described previously [21]. Four clones were sequenced for each amplicon excised from the RISA gel.
Clone Library Construction
PCR clone libraries for partial 16S rRNA genes were constructed as follows. Briefly, 50 ng of total bacterial DNA was used as a template in a final reaction volume of 10 μl, including 3 pmol of each primer and 0.5 U of Ex Taq HS DNA polymerase (Takara Bio, Otsu, Japan). To minimize the amplification of the 16S rRNA gene of soybean chloroplasts (X06428), we chose endophyte-specific primer 799f (aac (a/c)gg att aga tac cc(g/t)) [8] and 1525R (aag gag gtg wtc car cc) [27]. Cycling conditions were: initial denaturation for 3 min at 95°C; then 30 cycles consisting of 20 s at 94°C, 40 s at 53°C, and 2 min at 72°C; and a final extension for 10 min at 72°C. PCR products were resolved by 1% agarose gel electrophoresis in 1× TAE buffer. PCR products of the predicted size (700 to 800 bp) were extracted from the gels by using a QIAquick Gel Extraction Kit (Qiagen, Tokyo, Japan) and ligated into the plasmid vector pT7blue at 16°C overnight by using a DNA ligation kit, Mighty Mix (Takara Bio). ElectroTen-Blue Electroporation-Competent Cells (Stratagene, La Jolla, CA, USA) were then electroporated with the ligated DNA by using a Gene Pulser (Bio-Rad Laboratories, Tokyo, Japan). After the transformants had been cultured overnight at 37°C on Luria–Bertani agar plates containing ampicillin (50 µg/ml), 384 colonies were randomly selected from each library for sequencing. Sequencing analysis was conducted with a Type 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) using a BigDye Terminator Cycle Sequencing Reaction Kit (Applied Biosystems). Template DNAs were prepared by using an Illustra TempliPhi DNA Amplification Kit (GE Healthcare, Uppsala, Sweden). Both strands of the sequences were analyzed for the clone library by using the T7 (aat acg act cac tat ag) and kFw (ggg ttt tcc cag tca cga c) primer sets [48]. Sequences were edited with sequence assembly software (ATGC ver. 5.0; Genetyx, Tokyo, Japan) to remove the vector backbone, primer regions, and ambiguous sequences. After we had manually inspected the quality of the sequences, consensus sequences (approximately 700 bp) were generated by using sequence data from both ends of the clone.
Sequence Analysis
The sequences were analyzed for the presence of chimeras by using the CHIMERA_CHECK ver. 2.7 program of the Ribosomal Database Project (RDP)-II release 8.1 [9] and MALLARD [4]. Sequences identified as chimeric in both analyses were discarded, as were non-16S rDNA sequences. The remaining sequences were aligned by using CLUSTAL_X [54]. On the basis of the alignment, a distance matrix was constructed by using the DNADIST program from PHYLIP (ver. 3.66) [13] with the default parameters. The resulting matrices were used as input for DOTUR [49] to generate rarefaction curves, diversity indices, and species richness indicators. The default DOTUR settings were used, with threshold values of 97% sequence identity for the definition of operational taxonomic units (OTUs). Library coverage was calculated with the non-parametric estimator C [16], as described in Kemp and Aller [25]. The reciprocal of Simpson’s index (1/D) was used as a measure of diversity to evaluate the level of dominance in a community [61]. A DOTUR-formatted *.list file was used as input for SONS [50] in order to calculate the number of shared OTUs between libraries.
The phylogenetic composition of the sequences in each library was evaluated by using the Classifier program of RDP-II release 9.6 with confidence levels of 80% [59]. BLASTN [1] was also used to classify the clones and identify the nearest relatives in the GenBank database.
Phylogenetic Analysis
For the phylogenetic analysis, the reference sequences most closely related to the representative sequences of each OTU in the present study were searched in and downloaded from the Sequence Match program of RDP-II release 9.6 [9]. Sequences were aligned using the CLUSTAL W program [55]. The neighbor-joining method was used for building the trees [47]. The Phylip format tree output was applied by using the bootstrapping procedure [12]; 1,000 bootstrap trials were used. The trees were constructed with TreeView software [38].
Accession Numbers of Nucleotide Sequences
Nucleotide sequences of 16S rRNA genes for the PCR clone libraries and the amplicons of RISA profiles have been deposited in the DDBJ database under accession numbers AB459664–AB460995 (AB459664–AB459664 for library EB, AB459925–AB460252 for library CDB, AB460253–AB460528 for library CEB, AB460529–AB460754 for library CEN, AB460755–AB460995 for library CEP) and AB461321–AB461357, respectively.
Results
Enrichment of Bacterial Cells from Soybean Stems
The concentrations (0.1% to 1%) of Triton X-100 in the Tris-based extraction buffer were examined for the efficiency of disruption of chloroplast during the bacterial cell enrichment, by checking the color of the supernatant after differential centrifugations and by assessing the thickness of green color in the interface after the Nycodenz density gradient centrifugation, speculating that less green color of the supernatant and the interface is indicative of less contamination of plastid in the bacterial fraction. In the established protocol, the greenish colored plastids were almost completely removed after the final purification step with the Nycodenz density gradient centrifugation (Supplementary Fig. 1A). Microscopic observation revealed the presence of substances similar to bacterial aggregates in the enriched samples (Supplementary Fig. 1B). Approximately 2 μg of genomic DNA was obtained from 100 g of soybean stems. The plate counts of viable bacterial cells on NA medium were 7.2 × 105 and 6.4 × 104 cells/g (fresh weight of the stem) for pulverized stem powder and the enriched bacterial fraction, respectively.
The abundance and diversity of bacteria were semi-quantitatively evaluated by bacterial RISA. Both the intensity and the number of amplicons in bacterial RISA were much greater with the enriched samples than with the control DNA directly extracted from soybean stems (Fig. 2). Furthermore, sequencing analysis of these RISA amplicons showed that the major amplicons in the enriched samples were derived from bacteria, mostly well-known plant-associated bacteria (Supplementary Table 1). These results indicated that the enrichment method developed here was effective for extracting, purifying, and enriching bacterial cells from a plant tissue.

RISA profiles of stem-associated bacteria in soybeans. Lane M MapMarker 1000 (BioVentures, Murfreesboro, TN, USA); lanes 1 to 3 PCR products from the total DNA directly prepared from soybean stems pulverized by liquid nitrogen; lanes 4 to 6 PCR products from the total DNA of the bacterial fraction enriched from the stems. The leftmost numbers indicate the fragment lengths, and small letters on the right indicate the corresponding clone type in Supplementary Table 1
Statistics and Rarefaction Analysis of Clone Libraries
In total, five clone libraries were constructed in accordance with the flow chart shown in Fig. 1. We also initially attempted to construct a clone library using DNA sample directly extracted from stems pulverized in liquid nitrogen without the bacterial cell enrichment. However, most of the PCR amplicons in this library were exclusively derived from the 16S rRNA gene of soybean chloroplasts and other anonymous soybean genomic DNA (cross box in Fig. 1). Therefore, we omitted this clone library for further analysis.
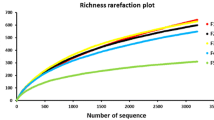
The respective clone libraries contained between 11 and 46 OTUs (defined by 97% sequence identity; Table 1). As expected, the highest value of OTU numbers was observed for the library EB. When all clones (n = 1,332) were combined, 757 unique sequences (data not shown) with a total of 72 OTUs were obtained (Table 1). The coverage of the clone libraries was at least 95% for all libraries, suggesting that our sampling effort from each library approached the total number of unique sequences within these libraries. Rarefaction curve showed that the library EB had the highest level of phylotype richness among libraries constructed (Fig. 3). Rarefaction curves for the libraries of epiphyte and endophyte (libraries CEP and CEN) were much closer to saturation, whereas the curves for the libraries EB, CEB, and CDB were not fully saturated.

Rarefaction curves of observed bacterial phylotype richness in clone libraries. OTUs were defined at 97% sequence identity by using DOTUR. The names of libraries stand for enriched bacteria (EB), cultured enriched bacteria (CEB), cultured directly prepared bacteria (CDB), epiphytic bacteria (CEP), and endophytic (CEN) bacteria, respectively
Diversity Indexes
The potential species richness was calculated for each library by using the non-parametric estimators Chao1 and ACE (Table 1). Both indexes yielded similar OTU estimations for all libraries. Calculations of OTU numbers, richness estimates, and Shannon’s and Simpson’s indexes for community diversity showed that the library EB had a higher richness and diversity than the rest of the libraries (Table 1). While these indexes were the lowest for the library CEN.
Phylogenetic Compositions of Clone Libraries
In order to assess the potential bias of the cell enrichment method for the bacterial diversity, the phylogenetic composition of the library EB was compared with other libraries derived from the culturable fractions of enriched bacterial cells (library CEB), pulverized stems (library CDB), and culturable epiphytic/endophytic bacteria (libraries CEP and CEN). Using RDP Classifier, the sequences obtained in clone libraries were placed into a taxonomic hierarchy. The relative abundances of the main phyla, as determined at the 80% confidence level, are shown in Table 1. Most of soybean stem-associated bacteria were classified into four bacterial phyla, the Bacteroidetes, Firmicutes, Actinobacteria, and Proteobacteria, except seven clones, which were classified as unassigned bacterial groups in current bacterial taxonomy. Although the most abundant phylum throughout all libraries was Proteobacteria (comprising 85% of clones in all libraries combined), the taxonomic distributions for each library were unique, even at the phylum or class level. Notably, the taxonomic distributions of clones in libraries EB and CEB were very similar, consistent with the fact that library CEB was derived from a culturable portion of the library EB (Fig. 1).
In contrast, 97% of clones in the library CDB were classified into Gammaproteobacteria. The library CEP contained 53.5% of clones for Proteobacteria and 37.3% for Bacteroidetes; the high abundance of Bacteroidetes was due to the presence of Sphingobacteria-like clones (comprising 34.9% in this library), which RDP Classifier classified as “unclassified Sphingobacteriaceae” (Fig. 4). Of the library CEN, 99% were shown to be Proteobacteria (82.7% and 16.4% for Alpha- and Gammaproteobacteria, respectively). The phyla Bacteroidetes and Betaproteobacteria were not detected in the library CEN.

Phylogenetic distribution of representative sequences in the clone libraries of 16S rRNA genes. The dendrogram (left) indicates the phylogenetic relationships among the representative sequences of OTUs. The table indicates the numbers of clones belonging to each OTU in each library and the results of the BLAST search using the representative sequences for each OTU. The sequences discussed in the main text are highlighted with gray background
Phylogenetic Diversity of Clone Libraries Assessed by Clustering Sequences at 97% Identity
The abundance of 16S rRNA gene clones in each library was assessed on the basis of the 72 OTUs defined at 97% sequence identity (Fig. 4). Among the 72 OTUs, 57 were covered by clones derived from the libraries EB and CEB, and 49 were represented only by the library EB (Fig. 4), suggesting that the more diverse bacteria were obtained in the enriched bacterial fraction. It was also clear that most of OTUs in libraries CEP and CEN (82.4% and 72.7%, respectively) were overlapped with the library EB (Fig. 4), indicating that both epiphytic and endophytic bacteria were fairly extracted from soybean stems by the bacterial cell enrichment method developed.
The most abundant clones throughout all libraries were shown to be Agrobacterium sp. (Alphaproteobacteria) represented by OTU AP7 consisting of 390 clones (29.3% of total number of clones) (Figs. 4 and 5). In particular, this OTU was dominant in the libraries CEB and CEN (55.8% and 78.8%, respectively) (Fig. 4). The second largest OTU was GP10 that of Enterobacter sp. (Gammaproteobacteria) (Figs. 4 and 5), which accounted for 64.3% in the library CDB. Although the overall distributions of taxonomic groups in libraries EB and CEB were very similar at phylum levels, there were some noticeable differences at the OTU level. The most substantial difference was the abundance for OTU AP7, comprising 6.5% and 55.8% in libraries EB and CEB, respectively. On the other hand, the OTU AP5 was exclusively presented and highly abundant in the library EB (19.5%), which was not detected in the library CEB. Another characteristic of the library EB was to cover a taxonomically wider range of Actinobacteria, including three suborders (Frankineae, Propionibacterineae, and Micrococcineae) (Figs. 4 and 5), than is found in other culturable libraries (CEB, CDB, CEP, and CEN).

Phylogenetic tree of soybean stem-associated bacteria based on the representative sequences of OTUs in clone libraries analyses of 16S rRNA genes. The tree was constructed by the neighbor-joining method. The scale represents 0.1 substitutions per site. The numbers at the nodes are the proportions of 1,000 bootstrap resamplings, and values of <500 are not shown. The OTUs and accession numbers of representative sequences determined in the present study are indicated in bold
Meanwhile, the dominance of Gammaproteobacteria (97%) in the library CDB was mainly explained by only three OTUs (OTU GP2 in Pseudomonadales [17.7%%], OTU GP10 in Enterobacteriales [64.3%], and OTU GP14 in Xanthomonadales [10.7%] in Fig. 4). Among them, the most dominant OTU GP10 (64.3%) (Enterobacter sp.) was shown to be the highest abundance as an OTU in a library in the present study, and was only found in the library CDB. The high abundance of Bacteroidetes in the library CEP was due to the presence of OTU BA1 which was a group of Sphingobacterium-like species. In the present study, the library CEN had the most biased phylogenetic composition, caused by OTU AP7 (Agrobacterium sp.) comprising 78.8% in the library CEN.
A phylogenetic tree was constructed by using the representative sequences of the 72 OTUs (Fig. 5). The result clearly indicated that soybean stems support a highly complex bacterial community including a series of well-known plant-associated bacterial groups such as Alpha- and Betaproteobacteria and Actinobacteria in addition to Gammaproteobacteria.
Discussion
In order to eliminate the interference of plant DNA in the bacterial community analysis, we developed a bacterial cell enrichment technique. Stem samples were homogenized with a buffer in a blender, and large plant debris and insoluble starch particles were removed by filtration through Miracloth followed by differential centrifugation. While Triton X-100 is considered as the most efficient detergent for disrupting the membrane system of chloroplast [10], this detergent is considered to be milder and less destructive to bacterial cells than SDS and other ionic detergents. This is of particular note because chloroplast DNA is a major disturbance in the 16S rRNA gene analyses of plant-associated bacteria by means of culture-independent methodologies [8]. The Nycodenz density gradient centrifugation has been used for concentrating and purifying animal cells due to the low cellular toxicity [44], and has been employed to extract bacterial cells from a soil matrix [30].
The bacterial cell enrichment method developed in the present study allowed us to conduct the 16S rRNA gene-based clone library analysis with soybean stems. Diverse bacterial sequences representing the major phyla of plant-associated bacteria were successfully amplified from the DNA prepared from the enriched bacterial cells (Figs. 4 and 5). As expected, the diversity of the enriched bacteria (library EB) was clearly higher than those from three conventional culturable bacterial fractions from plant powder (library CDB), epiphytic (library CEP), and endophytic bacteria (library CEN) (Table 1).
The culturability of the enriched bacteria was also evaluated. The results indicated that the taxonomic compositions were comparable between libraries EB and CEB at the levels of phylum to genus (Table 1 and Fig. 4). Furthermore, the taxonomic coverage for the culturable fraction of the enriched bacteria was also wider than those for the bacteria prepared by conventional culture methods (libraries CDB, CEP, and CEN) (Fig. 4), suggesting that the enriched method would be useful not only for the culture-independent analyses, but also for the culture-dependent analyses of plant-associated bacteria. Besides the assessment of the bacterial diversities for the enriched fraction, the comparisons between the libraries CEP and CEN provided an insight for the spatial distribution of stem-associated bacteria. Thus, there were biased distributions for several OTUs in the libraries CEP (OTUs BA1, BP6 FM1, and GP1 in Fig. 4) and CEN (OTUs AP7 and GP2 in Fig. 4). Particularly, the biased distribution of the Sphingomonas sp. for the library CEP was evident. These results would be useful for elucidating the spatial heterogeneity for the diversity of plant-associated bacteria.
By culture-independent methods, the microbial communities of the aerial part of plant tissues have been far less analyzed compared to those of rhizosphere [2]. In fact, only two reports have been published on 16S rRNA gene clone library-based community analysis of stem-associated bacteria in Thlaspi goesingense [19] and poplar [57] by using universal bacterial primers. When we compared our results with these previous two studies, the overall structures of stem-associated bacteria were very similar at the levels of phylum or class, despite the difference of plant species. The Proteobacteria (Alpha-, Beta-, and Gammaproteobacteria) was the most dominant group in all three studies, and the remaining groups consisted mainly of Actinobacteria, Firmicutes, and Bacteroidetes. It has been reported that the dominant isolates of plant endophytic bacteria belong to the genera of Pseudomonas, Bacillus, Enterobacter, and Agrobacterium [17], and that endophytic bacteria isolated from legume plants include Aerobacter, Agrobacterium, Bacillus, Chryseomonas, Curtobacterium, Enterobacter, Erwinia, Pseudomonas, and Sphingomonas [14, 53]. Most of these bacterial groups, except for Bacillus spp., were found in our clone library analysis. The high abundance of Agrobacterium sp. observed in the present study was also reported in a previous work for bacterial endophytes [15].
Meanwhile, the results in the present study were in contrast to a previous study which reported the dominance of Gammaproteobacteria in the soybean phytosphere [28]. We also have observed the dominance of Gammaproteobacteria in a culture of endophytic bacteria of soybean stems which were comparable materials in the present study [37]. In the present study, while the strong bias toward to Gammaproteobacteria was also observed for the bacterial diversity with the pulverized stems (library CDB), the analyses of enriched bacterial diversity revealed that soybean stems accommodate a wide variety of other bacterial groups, including Alphaproteobacteria as a dominant taxon (libraries EB and CEB). These results suggested that the bacterial cell enrichment would reflect the less biased diversity of plant-associated bacteria compared to the diversity described by conventional culture-dependent methods. The bacterial diversity of the enriched fraction should be examined with a fresh plant material in a future work, since it may be higher than with a frozen materials in the present study. The reason of strong bias toward to Gammaproteobacteria in culturing soybean-associated bacteria by conventional methods remains to be examined.
Leguminous plants control the degree of nodulation and mycorrhization of roots by rhizobia and mycorrhizae, respectively [7, 33, 36]. However, the degree to which plants use similar or identical systems for interactions with other microorganisms remains unclear. Recently, it was shown that wild-type and nodulation mutants of the model legume Medicago truncatula possess different bacterial community structures [35]. Also, we have demonstrated the significant impact of nodulation phenotypes on the microbial community in the rhizosphere [20]. Therefore, the bacterial enrichment procedures described here would be a useful tool to elucidate the impacts of the symbiotic signaling pathways and autoregulation on microbial communities in the aerial parts of plants, including the endosphere and the phyllosphere.
Leveau [29] has recently pointed out the significance of metagenomics for the study of plant growth-promoting bacteria. To attain the goal, further technical development is required for bacterial cell enrichment procedures by physical, chemical, or biological means [29]. The potential for metagenomics of plant-associated bacterial communities may lead to the discovery of novel plant growth-promoting genes, and the characterization of (not-yet-) culturable microbes in the phytosphere [29].
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Andrews JH, Harris RF (2000) The ecology and biogeography of microorganisms on plant surfaces. Annu Rev Phytopathol 38:145–180
Araújo WL, Marcon J, Maccheroni WJ, Van Elsas JD, Van Vuurde JWL, Azevedo JL (2002) Diversity of endophytic bacterial populations and their interaction with Xylella fastidiosa in citrus plants. Appl Environ Microbiol 68:4906–4914
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ (2006) New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72:5734–5741
Becker R, Behrendt U, Hommel B, Kropf S, Ulrich A (2008) Effects of transgenic fructan-producing potatoes on the community structure of rhizosphere and phyllosphere bacteria. Fems Microbiol Ecol 66:411–425
Cardinale M, Brusetti L, Quatrini P, Borin S, Puglia AM, Rizzi A, Zanardini E, Sorlini C, Corselli C, Daffonchio D (2004) Comparison of different primer sets for use in automated ribosomal intergenic spacer analysis of complex bacterial communities. Appl Environ Microbiol 70:6147–6156
Carroll BJ, McNeil DL, Gresshoff PM (1985) Isolation and properties of soybean [Glycine max (L.) Merr.] mutants that nodulate in the presence of high nitrate concentrations. Proc Natl Acad Sci U S A 82:4162–4166
Chelius MK, Triplett EW (2001) The diversity of Archaea and Bacteria in association with the roots of Zea mays L. Microb Ecol 41:252–263
Cole JR, Chai B, Marsh TL, Farris RJ, Wang Q, Kulam SA, Chandra S, McGarrell DM, Schmidt TM, Garrity GM, Tiedje JM et al (2003) The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res 31:442–443
Deamer DW, Crofts A (1967) Action of Triton X-100 on chloroplast membranes. Mechanisms of structural and functional disruption. J Cell Biol 33:395–410
Dent KC, Stephen JR, Finch-Savage WE (2004) Molecular profiling of microbial communities associated with seeds of Beta vulgaris subsp. Vulgaris (sugar beet). J Microbiol Methods 56:17–26
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Felsenstein J (1989) PHYLIP—phylogeny of inference package (version 3.2). Cladistics 5
Gagne S, Richard C, Roussean H, Antoun H (1987) Xylem-residing bacteria in alfalfa roots. Can J Microbiol 33:996–1000
Garbeva P, Overbeek LS, Vuurde JW, Elsas JD (2001) Analysis of endophytic bacterial communities of potato by plating and denaturing gradient gel electrophoresis (DGGE) of 16S rDNA based PCR fragments. Microb Ecol 41:369–383
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Hallmann J, Quadt-Hallmann A, Mahaffee WF, Kloepper JW (1997) Bacterial endophytes in agricultural crops. Can J Microbiol 43:895–914
Hung PQ, Kumar SM, Govindsamy V, Annapurna K (2007) Isolation and characterization of endophytic bacteria from wild and cultivated soybean varieties. Biol Fertil Soils 44:155–162
Idris R, Trifonova R, Puschenreiter M, Wenzel WW, Sessitsch A (2004) Bacterial communities associated with flowering plants of the Ni hyperaccumulator Thlaspi goesingense. Appl Environ Microbiol 70:2667–2677
Ikeda S, Rallos LEE, Okubo T, Eda S, Inaba S, Mitsui H, Minamisawa K (2008) Microbial community analysis of field-grown soybeans with different nodulation phenotypes. Appl Environ Microbiol 74:5704–5709
Ikeda S, Tsurumaru H, Wakai S, Noritake C, Fujishiro K, Akasaka M, Ando K (2008) Evaluation of the effects of different additives in improving the DNA extraction yield and quality from Andosol. Microbes Environ 23:159–166
Ikeda S, Watanabe KN, Minamisawa K, Ytow N (2004) Evaluation of soil DNA from arable land in Japan using a modified direct-extraction method. Microbes Environ 19:301–309
Jiao J-Y, Wang H-X, Zeng Y, Shen Y-M (2006) Enrichment for microbes living in association with plant tissues. J Appl Microbiol 100:830–837
Kadivar H, Stapleton AE (2003) Ultraviolet radiation alters maize phyllosphere bacterial diversity. Microb Ecol 45:353–361
Kemp PF, Aller JY (2004) Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. Fems Microbiol Ecol 47:161–177
Kent AD, Triplett EW (2002) Microbial communities and their interactions in soil and rhizosphere ecosystems. Annu Rev Microbiol 56:211–236
Kerkhof L, Santoro M, Garland J (2000) Response of soybean rhizosphere communities to human hygiene water addition as determined by community level physiological profiling (CLPP) and terminal restriction fragment length polymorphism (TRFLP) analysis. FEMS Microbiol Lett 184:95–101
Kuklinsky-Sobral J, Araujo WL, Mendes R, Geraldi IO, Pizzirani-Kleiner AA, Azevedo JL (2004) Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ Microbiol 6:1244–1251
Leveau JHJ (2007) The magic and menace of metagenomics: prospects for the study of plant growth-promoting rhizobacteria. Eur J Plant Pathol 119:279–300
Lindahl V, Bakken LR (1995) Evaluation of methods for extraction of bacteria from soil. Fems Microbiol Ecol 16:135–142
Lindow SE, Brandl MT (2003) Microbiology of the phyllosphere. Appl Environ Microbiol 69:1875–1883
Lodewyckx C, Vangronsveld J, Porteous F, Moore ERB, Taghavi S, Mezgeay M, Van der Lelie D (2002) Endophytic bacteria and their potential applications. Crit Rev Plant Sci 21:583–606
Meixner C, Vegvari G, Ludwig-Müller J, Gagnon H, Steinkellner S, Staehelin C, Gresshoff P, Vierheilig H (2007) Two defined alleles of the LRR receptor kinase GmNARK in supernodulating soybean govern differing autoregulation of mycorrhization. Physiol Plant 130:261–270
Normander B, Prosser JI (2000) Bacterial origin and community composition in the barley phytosphere as a function of habitat and presowing conditions. Appl Environ Microbiol 66:4372–4377
Offre P, Pivato B, Siblot S, Gamalero E, Corberand T, Lemanceau P, Mougel C (2007) Identification of bacterial groups preferentially associated with mycorrhizal roots of Medicago truncatula. Appl Environ Microbiol 73:913–921
Oka-Kira E, Kawaguchi M (2006) Long-distance signaling to control root nodule number. Curr Opin Plant Biol 9:496–502
Okubo T, Ikeda S, Kaneko T, Eda S, Mitsui H, Sato S, Tabata S, Minamisawa K (2009) Nodulation-dependent communities of culturable bacterial endophytes from stems of field-grown soybeans. Microbes Environ doi:10.1264/jsme2.ME09125
Page RDM (1996) TreeView: an application to display phylogenetic trees on personal computers. Bioinformatics 12:357–358
Pimentel IC, Glienke-Blanco C, Gabardo J, Stuart RM, Azevedo JL (2006) Identification and colonization of endophytic fungi from soybean (Glycine max (L.) Merril) under different environmental conditions. Braz Arch Biol Technol 49:705–711
Rasche F, Hödl V, Poll C, Kandeler E, Gerzabek MH, Van Elsas JD, Sessitsch A (2006) Rhizosphere bacteria affected by transgenic potatoes with antibacterial activities compared with the effects of soil, wild-type potatoes, vegetation stage and pathogen exposure. Fems Microbiol Ecol 56:219–235
Rasche F, Trondl R, Naglreiter C, Reichenauer TG, Sessitsch A (2006) Chilling and cultivar type affect the diversity of bacterial endophytes colonizing sweet pepper (Capsicum annum L.). Can J Microbiol 52:1036–1045
Reiter B, Sessitsch A (2006) Bacterial endophytes of the wildflower Crocus albiflorus analyzed by characterization of isolates and by a cultivation-independent approach. Can J Microbiol 52:140–149
Reiter B, Wermbter N, Gyamfi S, Schwab H, Sessitsch A (2003) Endophytic Pseudomonas spp. populations of pathogen-infected potato plants analysed by 16S rDNA- and 16S rRNA-based denaturating gradient gel electrophoresis. Plant Soil 257:397–405
Rickwood D, Ford T, Graham J (1982) Nycodenz: a new nonionic iodinated gradient medium. Anal Biochem 123:23–31
Saito A, Ikeda S, Ezura H, Minamisawa K (2007) Microbial community analysis of the phytosphere using culture-independent methodologies. Microbes Environ 22:93–105
Saito A, Kawahara M, Ikeda S, Ishimine M, Akao S, Minamisawa K (2008) Broad distribution and phylogeny of anaerobic endophytes of cluster XIVa clostridia in plant species including crops. Microbes Environ 23:73–80
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sato S, Kotani H, Nakamura Y, Kaneko T, Asamizu E, Fukami M, Miyajima N, Tabata S (1997) Structural analysis of Arabidopsis thaliana chromosome 5. I. Sequence features of the 1.6 Mb regions covered by twenty physically assigned P1 clones. DNA Res 4:215–219
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schloss PD, Handelsman J (2006) Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72:6773–6779
Seghers D, Wittebolle L, Top EM, Verstraete W, Siciliano SD (2004) Impact of agricultural practices on the Zea mays L. endophytic community. Appl Environ Microbiol 70:1475–1482
Sessitsch A, Reiter B, Peifer U, Wilhelm E (2002) Cultivation-independent population analysis of bacterial endophytes in three potato varieties based on eubacterial and Actinomycetes-specific PCR of 16S rRNA genes. Fems Microbiol Ecol 39:23–32
Sturz AV, Christie BR, Matheson BG, Nowak J (1997) Biodiversity of endophytic bacteria which colonize red clover nodules, roots, stems and foliage and their influence on host growth. Biol Fertil Soils 25:13–19
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tian X, Cao L, Tan H, Han W, Chen M, Liu Y, Zhou S (2007) Diversity of cultivated and uncultivated actinobacterial endophytes in the stems and roots of rice. Microb Ecol 53:700–707
Ulrich K, Ulrich A, Ewald D (2008) Diversity of endophytic bacterial communities in poplar grown under field conditions. Fems Microbiol Ecol 63:169–180
Wang HX, Geng ZL, Zeng Y, Shen YM (2008) Enriching plant microbiota for a metagenomic library construction. Environ Microbiol 10:2684–2691
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Yang CH, Crowley DE, Borneman J, Keen NT (2001) Microbial phyllosphere populations are more complex than previously realized. Proc Natl Acad Sci U S A 98:3889–3894
Zhou J, Xia B, Treves DS, Wu LY, Marsh TL, O'Neill RV, Palumbo AV, Tiedje JM (2002) Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68:326–334
Acknowledgements
This work was supported in part by Special Coordination Funds for Promoting Science and Technology, by Grant-in-Aid for Scientific Research (no. 17658034) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by a grant of Genomics for Agricultural Innovation (PMI-0002) from Ministry of Agriculture, Forestry and Fisheries of Japan.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
(gif 140 kb)
Supplementary Table 1
Sequence similarities of RISA amplicons to known species (gif 149 kb)
Rights and permissions
About this article
Cite this article
Ikeda, S., Kaneko, T., Okubo, T. et al. Development of a Bacterial Cell Enrichment Method and its Application to the Community Analysis in Soybean Stems. Microb Ecol 58, 703–714 (2009). https://doi.org/10.1007/s00248-009-9566-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-009-9566-0