
Abstract
Real-time polymerase chain reaction (PCR) is considered a highly sensitive method for the quantification of microbial organisms in environmental samples. This study was conducted to evaluate real-time PCR with SybrGreen detection as a quantification method for sulfate-reducing bacteria (SRB) in industrial wastewater produced by several chemical industries. We designed four sets of primers and developed standard curves based on genomic DNA of Desulfovibrio vulgaris from pure culture and on plasmids containing dissimilatory sulfate reductase (dsrA) or adenosine-5′-phosphosulfate reductase (apsA) genes of SRB. All the standard curves, two for dsrA and two for apsA genes, had a linear range between 0.95 × 102 and 9.5 × 106 copies/μL and between 1.2 × 103 and 1.2 × 107 copies/μL, respectively. The theoretical copy numbers of the tenfold dilutions of D. vulgaris genomic DNA were best estimated (between 2.7 to 10.5 times higher than theoretical numbers) by the standard curve with DSR1F and RH3-dsr-R primers. To mimic the effect of foreign DNA in environmental samples, serial dilutions of D. vulgaris genomic DNA were mixed with Escherichia coli chromosomal DNA (40 ng per assay). This influenced neither PCR amplification nor the quantification of target DNA. Industrial wastewater was sampled during a 15-month period and analyzed for the presence of SRB, based on dsrA gene amplification. SRB displayed a higher abundance during the summer (about 107–108 targets mL−1) and lower during the winter (about 104–105 targets mL−1). The results indicate that our real-time PCR approach can be used for detection of uncultured SRB and will provide valuable information related to the abundance of SRB in durable environmental samples, such as complex and saline industrial wastewaters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sulfate-reducing bacteria (SRB) are anaerobic microorganisms that use sulfate as an electron acceptor. They are known to grow both heterotrophically, using small organic molecules, and autrophically, using H2 as the electron donor and CO2 as the carbon source. However, several studies demonstrated that at least certain species of SRB do not only tolerate high concentrations of oxygen, but can also utilize oxygen as a terminal electron acceptor [8, 16]. SRBs are known to be present in the complex consortia of microorganisms involved in the anaerobic digestion processes used in municipal and industrial wastewater treatment.
A major drawback of sulfate reduction in wastewater treatments is the production of toxic H2S, which is also a possible precursor of odorants and significantly enhances microbially mediated corrosion of treatment facilities [32]. Furthermore, sulfate reduction may account for up to 50% of the mineralization of organic matter in aerobic wastewater treatment systems [19]. SRBs also have great economic impact in the oil industry, where they cause severe problems, including souring of oil and gas deposits and corrosion of production facilities [13, 30].
Considerable efforts have been directed toward the development of rapid and dependable methods for detection and enumeration of SRBs in natural and industrial environments. In general, the methods used to enumerate SRB can be divided into the following two categories: direct detection methods and culture methods. Conventional culture-based methods for studying SRBs are time consuming as a result of slow bacterial growth rates [26], and do not necessarily provide an accurate assessment of the types and numbers of bacteria present in a biological sample [15, 42]. The application of molecular biological methods to investigate the occurrence and distribution of bacteria in the environment has the advantage of providing direct information on community structure [7]. The direct detection methods developed recently include the use of antibodies raised against SRB [25], an immunoassay for the enzyme adenosine-5′-phosphosulfate (APS) reductase [31], and the use of 16S rRNA probes [2, 7]. Although promising, these techniques are still in the developmental phase and several problems are encountered when they are used in situ [3]. For example, rRNA fluorescent probes are difficult to use in sediments or industrial wastewater because of the high background autofluorescence of inorganic particles [27]. Furthermore, not all known types of dissimilatory sulfate reducers in environmental samples can be unequivocally identified with the RNA probes described previously [34, 43]. In addition, SRBs constitute a heterogeneous group, including members of several phyla and domains. Thus, it is not feasible to use the 16S rRNA gene as a general molecular marker [5, 17, 45].
The dsrAB genes, which encode the dissimilatory sulfate reductase, the key enzyme in dissimilatory sulfate reduction, can be used as a phylogenetic marker for identification of SRBs [17, 45]. These genes can be found in all known sulfate-reducing prokaryotes [47]. Thus, dsrAB from all sulfate-reducing lineages can be targeted by a single set of conserved primers or specific primers from variable regions of dsrAB.
In addition to dissimilatory sulfite reductase, sulfate-reducing microorganisms possess adenosine-5′-phosphosulfate (APS) reductase [33]. The genes for APS reductase, apsBA from bacteria and aprBA from archaea, encode subunits that appear to form a 1:1 alpha beta-heterodimer [12]. Both subunits of the APS reductase are highly conserved, and the APS reductase genes have been proposed as a useful phylogenetic marker. The absence of ambiguous nucleotides in sequences of all strains tested indicates that probably only one apsA gene copy is present in each strain [11]. ApsA- and DsrAB-based phylogenies from a subset of 42 sulfate-reducing microorganisms were compared and revealed that the overall tree topologies of ApsA- and DsrAB-derived analyses were similar [11].
The purpose of this study was to develop rapid method for quantification of sulfate-reducing microbial communities in a complex environment of evaporation ponds for a complex industrial wastewater. This was carried out by utilizing universal real-time PCR primers for dsrA and apsA genes, and allowed for rapid high throughput determinations of SRBs in wastewater.
Materials and Methods
Bacterial Strain and Growth Conditions
Reference strains Desulfovibrio vulgaris subsp. vulgaris (DSM 644) of validly described sulfate-reducing microorganisms were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) as lyophilized cells. D. vulgaris was grown in Medium 63 (desulfovibrio medium from http://www.dsmz.de/index.htm) in universal bottles, in an anaerobic chamber (atmosphere N2 80%; CO2 10%; H2 10%) at 30°C.
Determination of Sulfide
Samples from industrial wastewater evaporation ponds were tenfold diluted up to 10−9 of initial concentrations by filtrated (0.1 μm) industrial wastewater in universal anaerobic tubes with addition of Luria–Bertani broth (0.1%). The counting tubes were incubated in anaerobic chamber at 30°C in the dark for 1 month and checked every week for growth. During incubation, 5 mL subsamples were removed with syringe from the tubes and immediately added into test tubes containing 20 mL of 20% (w/v) zinc acetate to precipitate dissolved sulfide. Sulfide was determined by iodometriric method [6].
Nucleic Acid Extraction and Determination of DNA Concentrations
Total genomic DNA from wastewater samples was extracted from obtained pellets (30 mL samples), using the MoBio Power Soil DNA isolation kit (MoBio Laboratories, Inc., Solana Beach, CA) with the one modification: DNA bounded to the silica filter membrane was washed twice with solution C5. The purified DNA was eluted in 60 μL of solution C6 (MoBio Laboratories, Inc.) and stored at −20°C. Power Soil DNA isolation kit solutions C2 and C3 are very efficient in removing contaminating organic and inorganic matter. In addition, before real-time PCR amplification, DNA extracts were diluted with sterile double distillate water to overcome PCR inhibition of traces of humic and other substances that are found in industrial wastewater. Genomic DNA from D. vulgaris was obtained by the same method and DNA concentrations of samples were determined by ND-1000 UV–Vis Spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE., USA). The copy number was calculated on the basis of the measured DNA concentration (ng/μL), an average molecular mass of 660 for a basepair (bp) in a double-stranded DNA and the genome size of D. vulgaris subsp. vulgaris (3,570,858 bp; AE017285); [14]) or sizes of plasmids harboring dsrAB (4.9 kb) and apsA (3.9 kb).
PCR Amplification of dsrAB and apsA Gene Fragments
An ∼1.9-kb fragment encompassing parts of the dissimilatory sulfite reductase genes dsrA and dsrB and an ∼0.9-kb fragment of reductase α-subunit gene apsA from total genomic DNA, were amplified, using DSR1F and DSR4R [45] and APS7-F and APS8-R primers [11], respectively, with a Mastercycler gradient thermocycler (Eppendorf, Westbury, NY). Reaction mixtures included a 12.5-μl ReddyMix (PCR Master mix containing 1.5 mM MgCl2 and 0.2 mM concentration of each deoxynucleoside triphosphate; ABgene, Surrey, UK), a 1 pmol each of the forward and reverse primers, 1 to 2 μL of the sample preparation, plus water to bring the total volume to 25 μL. An initial denaturation-hot start of 4 min at 95°C was followed by 30 cycles of the following incubation pattern: 94°C for 40 s, 54°C for 40 s, and 72°C for 120 s (or 60 s for apsA fragment). The procedure was completed with a final elongation step at 72°C for 20 min.
Clone Library Construction and Sequencing
PCR heterologous products of approximately 1.9 kb dsrAB and 0.9 kb apsA were excised from agarose gels. The DNAs were purified from the gel slice by using the Wizard PCR Prep kit (Promega, Madison, WI). The purified PCR products were TA-cloned into the pGEM-T Easy vector, as specified by Promega (Madison, WI) and transformed into calcium chloride-competent XL MRF′ Escherichia coli cells, according to the manufacturer’s instructions. Plasmid DNA was isolated from individual clones by the Wizard Plus SV Minipreps DNA purification system (Promega). Aliquots from a subset of the samples of purified plasmid DNA were digested with EcoRI (MBI Fermentas) and then separated by electrophoresis on a 1% agarose gel. After staining with ethidium bromide, the bands were visualized on a UV transilluminator to select clones containing the appropriately sized insert. The clones with correct plasmid insert were then used for sequencing. Sequencing was performed by ABI PRISM dye terminator cycle sequencing ready reaction kit with an AmpliTaq DNA polymerase FS and DNA sequencer ABI model 373A system (PerkinElmer). One clone (208-49-2), harboring dsrAB, was fully sequenced using primers DSR1F and DSR4R [45] and sequencing primers DSR6F, DSR7F, DSR8F, DSR9F, and DSR10F [11] with the aim of using it in real-time PCR. The obtained sequence (1871 base) displayed 99% sequence identity to Desulfovibrio desulfuricans-G20 (CP000112) as best match by Mega Basic Local Alignment Search Tool (BLAST).
The sequences from appropriate libraries were aligned using ClustalW with the MEGA package: Molecular Evolutionary Genetics Analysis, version 3.1 package [20] and positions not sequenced in all isolates or with alignment uncertainties were removed. Phylogenetic trees were constructed by the neighbor-joining method [36] with the Mega package [20]. Bootstrap resampling analysis [9] for 100 replicates was performed to estimate the confidence of tree topologies.
Primer Design for Real-Time PCR
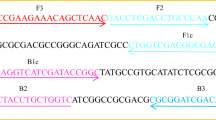
The universal primer pairs RH1-dsr-F/RH3-dsr-R and RH1-aps-F/RH2-aps-R (Table 1) were designed from conserved regions of multiple alignment of local (from industrial wastewater) dsrA or apsA sequences of SRBs (Fig. 2), obtained from constructed libraries, to generate specific DNA products of 164 and 191 bp within the dsrA and apsA genes, respectively (Fig. 5). In addition, another set of primers, DSR1F [45] with RH3-dsr-R and APS7-F [11] with RH2-aps-R, were used to amplify 222- and 279-bp fragments of the dsrA and apsA genes, respectively (Table 1).
All primers were checked by running a virtual PCR, and the amplifications were analyzed for expected product sizes, match (and mismatch) positions on each gene, and primer dimer formation, using a computer program (Amplify version 1.0; William Engles, Department of Genetics, University of Wisconsin, USA), and are displayed in Table 1. All primer sequences were examined by the BLAST to confirm the absence of significant homology with other known DNA sequences. The known sequences for dsrA and apsA from NCBI, and primers DSR1F [45] and APS7-F [11] and all newly designed primers underwent multiple alignment (Figs. 3 and 4) using ClustalW with the MEGA package: Molecular Evolutionary Genetics Analysis, version 3.1 package [20].
Real-Time PCR
Real-time PCR analysis was performed using the primers shown in Table 1. Quantification of bacterial DNA was performed in the ABI prism 7000 Sequence Detection System (Applied Biosystems), using Absolute QPCR SYBR Green ROX Mix (ABgene, Surrey, UK) in a 96-well optical plate. The PCR reaction consisted of 10 μL of Absolute QPCR SYBR Green ROX Mix, 150 nM each of forward and reverse primers, and 5.0 μL of each DNA template, in a total volume of 20 μL. Thermal cycling conditions were as follows: 2 min at 50°C, 15 min at 95°C, followed by 40 rounds of 15 s at 95°C and 1 min at 60°C. To verify that the used primer pair produced only a single specific product, a dissociation protocol was added after thermocycling, to determine dissociation of the PCR products from 60°C to 95°C. As standards for the calibration curves for quantification, D. vulgaris chromosomal DNA and plasmids with cloned 1.9 kb dsrAB and 0.9 apsA gene were used in six serial dilution points (in steps of tenfold). All runs included a no-template control. Detection was based on fluorescence resonance energy transfer, with a SYBR Green fluorophore. The cycle number at which the signal was first detected correlated with the original concentration of DNA template, whereas the starting copy number of amplicons was inversely proportional to the real-time threshold cycle (Ct). Standard curves were obtained by plotting the Ct value of each tenfold-dilution series of plasmids or genomic DNA of D. vulgaris. Plasmid standards and environmental samples were simultaneously assayed in duplicates and triplicates, respectively. Reproducibility of SYBR Green real-time PCR was assessed by running samples independently on different days. The PCR product was verified with ethidium-bromide-stained 2% agarose gels (SeaKemR LE Agarose; FMC BioProducts, Rockland, ME).
The ABI prism 7000 Sequence Detection System and SDS Software were used for data analysis. For data analyses, Ct values were exported into a Microsoft Excel Work sheet for further statistical analysis. The statistical significance of mean Ct values was determined and a P value of less than 0.05 was considered significant.
Nucleotide Sequence Accession Number
The sequences from this study have been deposited in the NCBI GenBank database under accession number EF052874 –EF052892 for dsrA sequences and EF052922–EF052936 for apsA sequences.
Results
Phylogenetic Relationships of dsrA and of apsA Gene Sequences
The phylogenetic analysis of 19 representative dsrA sequences that were retrieved from industrial wastewater evaporation ponds, showed that they were distantly related to known species of Desulfovibrio, Desulfomicrobium, and Desuldocella (with homology range between 85 and 92%). Some sequences showed higher homology (between 97 and 99%) to Desulfovibrio desulfuricans (Fig. 1A). The phylogenetic analysis of 15 representative apsA sequences showed them also distantly related to known species of Desulfovibrio and Desulfomicrobium (with homology range between 86 and 94%), except some sequences that exhibited the higher similarity (between 95 and 99%) to apsA of Desulfovibrio desulfuricans (Fig. 1B).

Phylogenetic relationships of dsrA gene sequences (A) and of apsA gene sequences (B) that were retrieved from industrial wastewater with members of related genera of SRB. The trees were constructed by the Neighbor–Joining method [36] with the Mega package [20] using partial sequences of dsrA and on apsA. The bar represents five substitutions per 100 nucleotide positions. Bootstrap probabilities [9] are indicated at branch nodes.
Specificity of PCR Primers
Highly specific PCR primer sets were developed for quantifying SRB from industrial wastewater. The conserved primer pairs RH1-dsr-F/RH3-dsr-R for dsrA and RH1-aps-F/RH2-aps-R for apsA (Table 1) were designed on multiple alignments of local dsrA or apsA sequences obtained from SRBs of industrial wastewater evaporation ponds (Fig. 2).

Multiple alignments of dsrA (A) and apsA (B) local sequences obtained from SRBs of industrial wastewater ponds used for designing conserved primer pairs RH1-dsr-F/RH3-dsr-R and RH1-aps-F/RH2-aps-R, respectively. Asterisks below the alignments indicate conserved positions.
Primers DSR1F [45] and APS7-F [11] and all newly designed primers were also aligned to dsrA (Fig. 3) and apsA (Fig. 4) sequences of wide-range bacterial genera of SRB to explore specificity of the primers. The DSR1F [45] and RH1-dsr-F forward primers display similar, but not identical specificity to different genera of SRB, where sequences of the region complementary to DSR1F are unknown or ambiguous (Fig. 3). The reverse primer RH3-dsr-R aligned consensus region and displayed high specificity to all genera of SRB (Fig. 3).

Sequence comparisons between universal PCR primers for dsrA and corresponding region of the genes from some genera of SRB. Dots indicate identical nucleotides and letters indicate nucleotide differences to those in the primers. No available data in nucleotide databases are represented by dashes. DSR-R primer suggested by Kondo et al. [18] are underlined.

Sequence comparisons between universal PCR primers for apsA and corresponding region of the genes from some genera of SRB. Dots indicate identical nucleotides and letters indicate nucleotide differences to those in the primers. No available data in nucleotides databases are represented by dashes. RH2-aps-R primer upper lined and sequence of APS-uni-F forward primer suggested by Friedrich, [11] embedded in sequence, which is underlined by thick line.
The APS7-F [11] and RH1-aps-F forward primers exhibit comparable specificity to different genera of SRB, but sequences of the region complementary to APS7-F are often unknown or ambiguous, whereas RH1-aps-F display higher specificity to some genera, for example, species of Desulfacinum (see Fig. 4). The reverse primer RH2-aps-R (Fig. 4 upperline) aligned consensus region especially of Desulfovibrio, Desulfonatronovibrio, Desulfohalobium, and Desulfomicrobium and displayed less specificity to all other genera of SRB.
The specificity of all four pairs of primers (Table 1) and the absence of unspecific products or primer dimers were tested by analyzing the real-time PCR reactions in 2% agarose gel stained with ethidium bromide. The molecular weight of the amplified product was as expected, and no other bands were visible (Fig. 5). Specific PCR products obtained from genomic DNA extracted from industrial wastewater samples were identified by SDS Software melting curve analysis (the negative derivative of fluorescence versus temperature, over the course of PCR product melt), and reproducible distinct melting points (Tm) of 87°C and 86°C were observed (data not shown) for the DSR1F/RH3-dsr-R (amplify 222 bp) and for RH1-dsr-F/RH3-dsr-R (amplify 164 bp) primers, respectively (Fig. 5; lanes 4 and 3). Additional two sets of primers, APS7-F/RH2-aps-R and RH1-aps-F/RH2-aps-R, for apsA gene amplified 279 and 191 bp amplicons (Fig. 5; lanes 2 and 1), generated specific peaks of Tm at 88°C and at 87°C, respectively (data not shown). Negative controls and amplification of DNA from non-SRB strain (E. coli) did not result in any PCR products.

Agarose (2.0%) gel of real-time PCR amplification products of dsrA and apsA genes with four sets of primers. Lane M—Marker DNA; lane 1 RH1-aps-F and RH2-aps-R primers (191 bp); lane 2 RH1-aps-F and RH2-aps-R primers (279 bp); lane 3 RH1-dsr-F and RH3-dsr-R primers (164 bp); and lane 4 DSR1F and RH3-dsr-R primers (222 bp)
Performance of Standard Curves
Using the plasmids harboring dsrA and apsA genes from local SRB strains, we made standard curves, by duplicate serial dilutions of known amounts of circular plasmid DNA. Linearity and reproducibility of the standard curves were tested using the primer sets (Table 1). The standard curve for dsrA gene with two sets of primers, DSR1F/RH3-dsr-R and RH1-dsr-F/RH3-dsr-R, had a linear range between 1.9 × 103 and 1.9 × 108 copies per assay (0.95 × 102 to 9.5 × 106 copies/μL) with a slope of −3.6 (R 2 value >0.99), and −3.4 (R 2 value >0.99), respectively (Fig. 6A). The standard curve for apsA gene with two sets of primers, APS7-F/RH2-aps-R and RH1-aps-F/RH2-aps-R, had a linear range between 2.4 × 104 and 2.4 × 108 copies per assay (1.2 × 103 to 1.2 × 107 copies/μL) with a slope of −4.0 (R 2 value >0.98), and −3.8 (R 2 value >0.98), respectively (Fig. 6B).

Standard curves for dsrA and apsA genes of SRBs using four sets of primers. (A) DSR1F/RH3-dsr-R (open circles), and RH1-dsr-F/RH3-dsr-R (open squares) for dsrA. (B) APS7-F/RH2-aps-R (open circles), and RH1-aps-F/RH2-aps-R (open squares) for apsA. Threshold cycle (Ct) was plotted against tenfold dilution of plasmid from 1.9 × 103 to 1.9 × 108 and 2.4 × 103 to 2.4 × 108 copies per assay for dsrA and apsA genes, respectively. Data points represent the average of duplicate measurements and error bars indicate the standard deviations.
Sensitivity and Detection Limits of dsrA and apsA in Desulfovibrio vulgaris Chromosomal DNA
To evaluate the sensitivity and detection limits of the primers, extracted DNA from pure culture of D. vulgaris was used in the real-time PCR assay. Plots of the calculated copy numbers against the threshold cycle (Ct) values obtained with the four primer sets resulted in a good linearity between 7.5 × 103 and 7.5 × 106 copies of D. vulgaris chromosome per assay (3.75 × 102 to 3.75 × 105 copies/μL). Correlation coefficients of curves for dsrA gene with two sets of primers, DSR1F/RH3-dsr-R and RH1-dsr-F/RH3-dsr-R, were R 2 value >0.99 (slope −3.93) and R 2 value >0.99 (slope −3.72), respectively (Fig. 7A). Correlation coefficients of curves for apsA gene with two sets of primers, APS7-F/RH2-aps-R and RH1-aps-F/RH2-aps-R, were R 2 value >0.99 (slope −3.80) and R 2 value >0.99 (slope −3.45), respectively (Fig. 7B).

Standard curves obtained by plotting the genomic DNA concentration of D. vulgaris using four sets of primers. (A) DSR1F/RH3-dsr-R (open circles), and RH1-dsr-F/RH3-dsr-R (open squares) for dsrA. (B) APS7-F/RH2-aps-R (open triangles), and RH1-aps-F/RH2-aps-R (open circles) for apsA. Relation between the threshold cycle (Ct) and the apparent amount of genomic DNA molecules of D. vulgaris (tenfold dilution from 7.5 × 103 to 7.5 × 106 copies per assay) obtained with SybrGreen real-time PCR. Data points represent the average of duplicate measurements and error bars indicate the standard deviations.
To determine whether the standard curves, nevertheless, give a good estimation of total dsrA or apsA gene copy numbers (one copy of each gene per genome), a serial dilution of D. vulgaris chromosomal DNA was used. Based on the measured DNA concentration of the sample, an average molecular mass of 660 for a base pair in a double-stranded DNA and the genome size of D. vulgaris subsp. vulgaris (3,570,858 bp; (AE017285)), we estimated the copy numbers in each of the dilutions (Table 2) for all four pairs of primers (Table 1). The theoretical copy numbers of the tenfold dilutions were best estimated by the DSR1F/RH3-dsr-R standard curve (2.7–10.5 times higher than theoretical numbers; Table 2). For the RH1-dsr-F/RH3-dsr-R standard curve, the obtained copy numbers were 15.7 and 80.6 times higher than the theoretical numbers. For the apsA gene standard curve, with the APS7-F/RH2-aps-R and RH1-aps-F/RH2-aps-R pairs of primers, the obtained copy numbers were from 0.71 to 5.5 and from 10.7 to 64.1 times higher than the theoretical number, respectively (Table 2).
To mimic the effect of foreign DNA in environmental samples, same serial dilutions of D. vulgaris genomic DNA were mixed with E. coli XL MRF′ (40 ng per assay). Measurement of the fractional number of D. vulgaris showed that foreign DNA does not particularly influence the quantification of target DNA (Table 2). Genomic DNA from D. vulgaris was also mixed with filtrated (0.1 μm) industrial wastewater and purified as mentioned (see “Materials and Methods”) and compared with extracted DNA from pure culture of D. vulgaris. No difference was observed in copy numbers of the tenfold dilutions (data not shown) that ensures our approach to overcome inhibition.
Real-Time PCR Quantification of SRB in Industrial Wastewater
In the current study, quantification of SRB was done in high-salinity industrial wastewater evaporation ponds. The ponds are the final treatment stage of a combined complex and saline wastewater mixture, contributed by several chemical plants at the Ramat-Hovav industrial park, in the Negev desert, Israel (manufacturing various pesticides, pharmaceuticals, aliphatic and aromatic halogens). Salt concentration in the pond at the time of sampling fluctuated between 12 to 16% (w/v). Organic matter concentration was 2–2.5 gC/L (on the basis of total organic carbon measure). The ponds receive a mixture of saline and high-strength industrial wastewater, creating a unique habitat for various microorganisms. Five industrial wastewater evaporation pools were analyzed for the presence of SRBs by real-time PCR, using SYBR Green I dye. The standard curve for dsrA (with DSR1F and RH3-dsr-R) primers was used. The measured values were transformed to targets per milliliter of wastewater. The level of SRBs displayed higher abundance during summer (June and July, about 108 targets mL−1) and lower during October to May 2005 (about 104–105 targets mL−1) and increased levels in July 2006 (about 5 × 106 targets mL−1; Fig. 8). Comparison of enumeration of SRB cell numbers by real-time PCR to most-probable-number (MPN) method (see “Materials and Methods”) revealed that the former technique (2.69 × 106 targets mL−1) was about 100-fold higher then latter (about 104 targets mL−1).

Seasonal evolution of SRBs in highly saline industrial wastewaters. The abundance of SRBs were evaluated at 6 m depth of the five industrial wastewater evaporation ponds and compared to the temperature in same depth. Bar heights represent means based on average samples of all five industrial wastewater ponds and error bars represent the standard deviations.
Discussion
Bacterial isolations and identifications are time consuming and prohibitive for large-scale studies. This is especially true when investigating the SRBs using culturing techniques, as there are requirements for samples to be processed quickly, as well as the need for specialized anaerobic techniques. However, current developments in molecular analysis have overcome many of these restrictions and the design and employment of carefully evaluated probes and primers is proving to be of value in identifying bacteria in complex microbial communities [1].
The majority of the dsrA and apsA sequences obtained from industrial wastewater ponds related to different species of Desulfovibrio, but sequences related to other genera of SRB such as Desulfomicrobium and Desulfocella were also revealed (Fig. 1). Most of the dsrA and apsA sequences that were retrieved from industrial wastewater ponds were found to be distantly related to known species except the sequences highly homologous to Desulfovibrio desulfuricans (Fig. 1). Therefore, it is possible that each local strain of SRB harbors the dsrA and apsA related to different species or genera of SRB. The highly saline industrial wastewaters have not been studied sufficiently for microbial diversity in general and on diversity of SRB in particular and might constitute a pool for novel strains of SRB.
The PCR primers were designed from multiple alignments of local (from industrial wastewater) dsrA or apsA sequences of SRBs (Fig. 2) obtained from constructed libraries. The RH1-dsr-F forward primer annealed conserved region, which lie downstream of about 50 bases from DSR1F [45] forward primer. These two primers are highly specific to more than a few genera of SRB. The RH1-dsr-F display low specificity to some species of SRB especially to Desulfotomaculum halophilum, Desulfotomaculum thermocisternum, Desulfotomaculum nigrificans (phylum Fermicutes), Desulfonema ishimotonii, Desulfofaba gelida, Desulfomusa hansenii, Desulfonatronum lacustre, Desulfoarculus baarsii, and Desulfovirga adipica (Fig. 3). Nevertheless, most of these sequences were deficient in region complementary to DSR1F. The RH3-dsr-R reverse primer aligned consensus region of all SRB genera used in Fig. 3 with high specificity. Our RH3-dsr-R primer is highly homologous to DSR-R primer suggested by Kondo et al. [18], but short by two bases on 3′-terminus and long by one base on 5′-terminus. DSR-R primer contains two additional guanine bases at the 3′-terminus (Fig. 3; the primer underlined), which may improve stability of the primer; however, these two bases display mismatches for some sequences (Fig. 3 in this article and see Fig. 2 from Kondo et al. [18]). Mismatched nucleotides at the 3′-ultimate and 3′-penultimate positions have previously been shown to be detrimental to the amplification process [21, 38]. This is primarily because of the need for a perfect 3′ basepair to allow enzymatic synthesis rather than any thermodynamic effect on duplex formation. Recently, it was shown that primers with substitution of nucleotide at the 3′-termini by inosine might overcome this limitation and richness cloned libraries for subsequent higher microbial diversity of studied environment [4].
The APS7-F [11] and RH1-aps-F forward primers exhibit comparable specificity to different genera of SRB, with some preference to RH1-aps-F (for example Desulfacinum and Desulfotomaculum; Fig. 4). Moreover, some sequences are deficient or ambiguous in the region complementary to APS7-F and it is unfeasible to compare specificity of the primers. The reverse primer RH2-aps-R (Fig. 4, upper line) aligned consensus region especially of species belonging to Desulfovibrio, Desulfonatronovibrio, Desulfohalobium, and Desulfomicrobium, whereas there were mismatched sequences of other genera from three to nine bases at the 3′-terminus (Fig. 4). Hence, the shortening of the sequence of RH2-aps-R and extension at the 5′-terminus (thick underlined) may substantially improve the primer universality. Nearly all of the sequence (Fig. 4; thick underlined) was originally used as forward primer (APS-uni-F) for sequencing of PCR products of apsA genes [11]. Multiple alignment of apsA revealed consensus region signed as RH3-aps-F primer (Fig. 4), which is more conserved than RH1-aps-F region. RH3-aps-F and sequence with a thick underline might be proposed as batter set of primers for PCR amplification (155 bp) of apsA genes.
The PCR primers developed in this article specifically detect and quantify SRBs in industrial wastewater. The method provides a highly sensitive means of identifying and quantifying these organisms and facilitates high throughput bacterial analysis of environmental samples. Industrial wastewater contains large microbial diversity, as defined by 16S rDNA libraries [4]. Real-time PCR analysis with dsrA and apsA primers had no secondary peaks (Fig. 5), indicating no nonspecific products. A good linear correlation was found between different concentrations of the plasmid-derived dsrA and apsA amplicons (Fig. 6) and genomic DNA of D. vulgaris (Fig. 7).
To determine and compare whether the standard curves (for each of all four sets of primers; Table 1) provide a good estimation of total dsrA or apsA gene copy numbers, a serial dilution of D. vulgaris chromosomal DNA was used (Table 2). The theoretical copy numbers of D. vulgaris were best estimated by the DSR1F/RH3-dsr-R standard curve (2.7–10.5 times higher) and less for the RH1-dsr-F/RH3-dsr-R standard curve (15.7 and 80.6 times higher). For the apsA gene standard curve, with the APS7-F/RH2-aps-R and RH1-aps-F/RH2-aps-R pairs of primers, the obtained copy numbers were from 0.71 to 5.5 and from 10.7 to 64.1 times higher than the theoretical number, respectively (Table 2). Annealing stability (as a function of GC content, number of mismatches, and primer length), secondary structure of the primers and its concentrations and different melting temperature of the primers of particular pair might have an effect on the absolute copy numbers for the same sample. These differences may be caused by the difference in slope of the standard curve and, thus, to the difference in amplification efficiency between the D. vulgaris DNA dilution series and the respective standard curves. Moreover, efficiency of different Tag DNA polymerases, concentrations of MgCl2, and some concentrations of SyberGreen dye (inhibits PCR to some extent) should be calibrated for each pair of primers to establish an adaptable real-time PCR protocol [40, 41]. Real-time PCR quantification of some genera of SRB with 16S rRNA gene primers can be overestimated by over 100-fold [39]. With specific primers for functional genes (dsrA and apsA), they may be overestimated from 0.7 up to 80-fold depending on the standard curve (and set of primers) used and the magnitude of initial copy number (Figs. 6 and 7; Table 2). Therefore, to ensure that all of the above should be taken into consideration to ensure specific amplification and subsequent accurate enumeration of the targets by real-time PCR, we chose RH1-dsr-F/RH3-dsr-R pair with the range of 2.7–10.5 times higher only than theoretical numbers.
Highly specific primers against 16S rDNA were previously designed for particular species of SRBs [22, 39–41]. Despite this, real-time PCR results cannot be directly correlated to cell numbers because 16S rRNA gene operons are not equal in all bacteria [44]. In addition, the heterogeneity found in conserved regions of 16S rRNA genes [10], plus the fact that some SRB strains harbor heterogenic sequences, which may vary between two to four types per strain [39], limits the utility of using 16S rDNA primers and is not adaptable for total SRB community monitoring.
The dsrAB and apsBA genes encode the dissimilatory sulfate reductase and adenosine-5′-phosphosulfate (APS) reductase, respectively. These are the key enzymes in dissimilatory sulfate reduction [33]. The dsrAB and apsBA genes can be found in all known sulfate-reducing prokaryotes and can be targeted by a single set of conserved primers from each of the specific primers from variable regions of the genes. These genes can therefore be used as phylogenetic markers for culture independent analyses of complex microbial communities to assess the potential for in situ biological sulfide production through anaerobic sulfate respiration by SRB [28]. Recently, a real-time reverse transcription (RT)-PCR method was developed (cDNA quantification by TaqMan probe) for the quantification of dissimilatory (bi)sulphite reductase mRNA in Desulfobacterium autotrophicum a as metabolic indicator of SRB, which also has the potential to be applied in natural environments [29]. This may prove to be an important method for discerning activity of SRBs, but less for quantification of the bacterial population.
Competitive PCR (cPCR) has been recently developed, based on the amplification of dsrAB genes [23, 24]. This method does not require a conserved internal sequence, but is more time-consuming [35] than the method presented in this article. It is based on the co-amplification of a target sequence and a standard internal sequence known as a competitor [46]. The main advantage of this technique is that the contaminants co-extracted with nucleic acids have similar effects on the amplification of the target and competitor sequences. The disadvantages of the technique include increased repeats required for verification purposes and a semiquantitative estimate based on gel electrophoresis. Real-time PCR has been previously used, as it overcomes the shortcomings of conventional PCR, and has an increased ability to quantify the number of gene copies present in the initial sample, often to one genome copy per microliter of DNA extract [37].
Using competitive PCR of the dsrAB genes Leloup et al., [24] ascertained the abundance of SRBs in a mudflat. They demonstrated that SRBs display seasonal dynamics with higher abundance and activity during the early summer. In addition, SRBs appeared to be mainly controlled by physical–chemical parameters (temperature and dissolved organic carbon concentration) and topographic evolution of the mudflat (erosion/deposit erosion) [24]. Our results showed that in the complex environment of highly saline and concentrated evaporation ponds, the level of SRBs demonstrated temperature dependence with higher abundance during summer 2005 (about 107–108 targets per mL with a temperature range from 28 to 31°C) and lower in autumn–spring (about 104–105 targets per mL, with temperature range from 16 to 27°C) (Fig. 8). During July of (summer) 2006 there was an increase in the abundance once more (about 5 × 106 targets mL−1, with an average temperature of about 28°C). The lower level of SRB detected during summer 2006, compared with the summer of 2005, may have been caused by differences in flow and diminished organic compounds in the wastewater during 2006. Enumeration of SRB cell numbers by real-time PCR was about 100-fold higher than most-probable-number (MPN) based on sulfide detection. Enumerations based on culture-based methods and standard MPN methods appear to underestimate cell numbers of SRB by at least a factor of 1000 and 10, respectively (for review, see reference [18]). Capability of the PCR methods used to count and distinguish between live and dead SRB should also be taken into consideration. Although results indicated in this article display high variability, this appears to be caused by proliferation of SRB rather than preservation ability of dead bacteria from high-molecular-weight DNA degradation. The results indicate that our real-time PCR approach can be used for detection of uncultured SRBs and will provide valuable information about the abundance of SRB in durable environmental samples such as highly saline industrial wastewaters.
References
Alm, EW, Oerther, DB, Larsen, N, Stahl, DA, Raskin, L (1996) The oligonucleotide probe database. Appl Environ Microbiol 62: 299–306
Amann, RI, Stromley, J, Devereux, R, Key, R, Stahl, DA (1992) Molecular and microscopic identification of sulfate-reducing bacteria in multispecies biofilms. Appl Environ Microbiol 58: 614–623
Amann, RI, Ludwig, W, Schleifer, KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59: 143–169
Ben-Dov, E, Shapiro, OH, Siboni, N, Kushmaro, A (2006) Advantage of using inosine at the 3′ termini of 16S rRNA gene universal primers for the study of microbial diversity. Appl Environ Microbiol 72: 6302–6306
Castro, HF, Williams, NH, Ogram, A (2000) Phylogeny of sulfate-reducing bacteria. FEMS Microbiol Ecol 31: 1–9
Clesceri, LS, Greenberg, AE, Eaton, AD (Eds.) (1998) Standard Methods for the Examination of Water and Wastewater, 20th edn., APHA, Washington, DC
Daly, K, Sharp, RJ, McCarthy, AJ (2000) Development of oligonucleotide probes and PCR primers for detecting phylogenetic subgroups of sulfate-reducing bacteria. Microbiology 146: 1693–1705
Dilling, W, Cypionka, H (1990) Aerobic respiration in sulfate-reducing bacteria. FEMS Microbiol Lett 71: 123–128
Felsenstein, J (1985) Confidence limits of phylogenies: an approach using the bootstrap. Evolution 39: 783–791
Forney, LJ, Zhou, X, Brown, CJ (2004) Molecular microbial ecology: land of the one-eyed king. Curr Opin Microbiol 7: 210–220
Friedrich, MW (2002) Phylogenetic analysis reveals multiple lateral transfers of adenosine-5′-phosphosulfate reductase genes among sulfate-reducing microorganisms. J Bacteriol 184: 278–289
Fritz, G, Buchert, T, Huber, H, Stetter, KO, Kroneck, PMH (2000) Adenylylsulfate reductases from archaea and bacteria are 1:1 alpha beta-heterodimeric iron-sulfur flavoenzymes-high similarity of molecular properties emphasizes their central role in sulfur metabolism. FEBS Lett 473: 63–66
Hamilton, WA (1985) Sulphate-reducing bacteria and anaerobic corrosion. Annu Rev Microbiol 39: 195–217
Heidelberg, JF, Seshadri, R, Haveman, SA, Hemme, CL, Paulsen, IT, Kolonay, JF, Eisen, JA, Ward, N, Methe, B, Brinkac, LM, Daugherty, SC, DeBoy, RT, Dodson, RJ, Durkin, AS, Madupu, R, Nelson, WC, Sullivan, SA, Fouts, DE, Haft, DH, Selengut, J, Peterson, JD, Davidsen, TM, Zafar, N, Zhou, L, Radune, D, Dimitrov, G, Hance, M, Tran, K, Khouri, HM, Gill, J, Utterback, TR, Feldblyum, TV, Wall, JD, Voordouw, G, Fraser, CM (2004) The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol 22: 554–559
Jain, DK (1995) Evaluation of the semisolid Postgate’s B medium for enumerating sulfate-reducing bacteria. J Microbiol Methods 22:27–38
Jonkers, HM, Koh, I-O, Behrend, P, Muyzer, G, de Beer, D (2005) Aerobic organic carbon mineralization by sulfate-reducing bacteria in the oxygen-saturated photic zone of a hypersaline microbial mat. Microbial Ecol 49: 291–300
Klein, M, Friedrich, M, Roger, AJ, Hugenholtz, P, Fishbain, S, Abicht, H, Blackall, LL, Stahl, DA, Wagner, M (2001) Multiple lateral transfers of dissimilatory sulfate reductase genes between major lineages of sulfate-reducing prokaryotes. Appl Environ Microbiol 67: 6028–6035
Kondo, R, Nedwell, DB, Purdy, KJ, de Queiroz Silva, S (2004) Detection and enumeration of sulphate-reducing bacteria in estuarine sediments by competitive PCR. Geomicrobiol J 21: 145–157
Kuhl, M, Jorgensen, BB (1992) Microsensor measurements of sulfate reduction and sulfide oxidation in compact microbial communities of aerobic biofilms. Appl Environ Microbiol 58: 1164–1174
Kumar, S, Tomura, K, Nei, M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5: 150–163
Kwok, S, Kellogg, DE, McKinney, N, Spasic, D, Goda, L, Levenson, C, Sninsky, JJ (1990) Effects of primer-template mismatches on the polymerase chain reaction: human immunodeficiency virus type 1 model studies. Nucleic Acids Res 18: 999–1005
Labrenz, M, Brettar, I, Christen, R, Flavier, S, Bötel, J, Höfle, MG (2004) Development and application of a real-time PCR approach for quantification of uncultured bacteria in the central Baltic sea. Appl Environ Microbiol 70: 4971–4979
Leloup, J, Quillet, L, Oger, C, Boust, D, Petit, F (2004) Molecular quantification of sulfate-reducing microorganisms (carrying dsrAB genes) by competitive PCR in estuarine sediments. FEMS Microbiol Ecol 47: 207–214
Leloup, J, Petit, F, Boust, D, Deloffre, J, Bally, G, Clarisse, O, Quillet, L (2005) Dynamics of sulfate-reducing microorganisms (dsrAB genes) in two contrasting mudflats of the seine estuary (France). Microbi Ecol 50: 307–314
Lillebæk, R (1995) Application of antisera raised against sulfate-reducing bacteria for indirect immunofluorescent detection of immunoreactive bacteria in sediment from the German Baltic Sea. Appl Environ Microbiol 61: 3436–3442
Macfarlane, GT, Gibson, GR (1991) Sulphate-reducing bacteria. In: Levett, PN (Ed.) Anaerobic microbiology—a practical approach. Oxford University Press, Oxford, pp 201–222
Muyzer, G, Ramsing, NB (1995) Molecular methods to study the organization of microbial communities. Water Sci Technol 32: 1–9
Nakagawa, T, Fukui, M (2003) Molecular characterization of community structures and sulfur metabolism within microbial streamers in Japanese hot springs. Appl Environ Microbiol 69: 7044–7057
Neretin, LN, Schippers, A, Pernthaler, A, Hamann, K, Amann, R, Jørgensen, BB (2003) Quantification of dissimilatory (bi)sulphite reductase gene expression in Desulfobacterium autotrophicum using real-time RT-PCR. Environ Microbiol 5: 660–671
Odom, JM (1990) Industrial and environmental concerns with sulfate-reducing bacteria. ASM News 56: 473–476
Odom, JM, Jessie, K, Knodel, E, Emptage, M (1991) Immunological cross-reactivities of adenosine-5′-phosphosulfate reductases from sulfate-reducing and sulfide-oxidizing bacteria. Appl Environ Microbiol 57: 727–733
Postgate, JR (1984) The sulphate-reducing bacteria, 2nd edn. Cambridge University Press, Cambridge, United Kingdom
Rabus, R, Hansen, TA, Widdel, F (1999) Dissimilatory sulfate- and sulfur-reducing prokaryotes. In: Dworkin, M, Schleifer, K-H, Stackebrandt, E (Eds) The prokaryotes: an evolving electronic database for the microbiological community. Springer, New York, pp 1–87
Ramsing, NB, Fossing, H, Ferdelman, TG, Andersen, F, Thamdrup, B (1996) Distribution of bacterial populations in a stratified fjord (Mariager Fjord, Denmark) quantified by in situ hybridization and related to chemical gradients in the water column. Appl Environ Microbiol 62: 1391–1404
Rayemaekers, L (2000) Basic principles of quantitative PCR. Mol Biotechnol 15: 115–122
Saito, N, Nei, M (1987) The neighbor-joining method: a new method for constructing phylogenetic trees. Mol Biol Evol 4: 406–425
Saleh-Lakha, S, Miller, M, Campbell, RG, Schneider, K, Elahimanesh, P, Hart, MM, Trevors, JT (2005) Microbial gene expression in soil: methods, applications and challenges. J Microbiol Methods 63: 1–19
Sarkar, G, Cassady, J, Bottema, CDK, Sommer, SS (1990) Characterization of polymerase chain reaction amplification of specific alleles. Anal Biochem 186: 64–68
Smits, TH, Devenoges, C, Szynalski, K, Maillard, J, Holliger, C (2004) Development of a real-time PCR method for quantification of the three genera Dehalobacter, Dehalococcoides, and Desulfitobacterium in microbial communities. J Microbiol Methods 57: 369–378
Stubner, S (2002) Enumeration of 16S rDNA of Desulfotomaculum lineage 1 in rice field soil by real-time PCR with SybrGreen detection. J Microbiol Methods 50: 155–164
Stubner, S (2004) Quantification of Gram-negative sulphate-reducing bacteria in rice field soil by 16S rRNA gene-targeted real-time PCR. J Microbiol Methods 57: 219–230
Tanner, RS (1989) Monitoring sulfate-reducing bacteria: comparison of enumeration media. J Microbiol Methods 10: 83–90
Teske, A, Wawer, C, Muyzer, G, Ramsing, NB (1996) Distribution of sulfate-reducing bacteria in a stratified fjord (Mariager Fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresis of PCR-amplified ribosomal DNA fragments. Appl Environ Microbiol 62: 1405–1415
Tourova, TP (2003) Copy number of ribosomal operons in prokaryotes and its effect on phylogenetic analysis. Microbiology 72: 437–452
Wagner, M, Roger, AJ, Flax, JL, Brusseau, GA, Stahl, DA (1998) Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J Bacteriol 180: 2975–2982
Zachar, V, Thomas, RA, Goustin, AS (1993) Absolute quantification of target DNA: a simple competitive PCR for efficient analysis of multiple samples. Nucleic Acids Res 21: 2017–2018
Zverlov, V, Klein, M, Lücker, S, Friedrich, MW, Kellermann, J, Stahl, DA, Loy, A, Wagner, M (2005) Lateral gene transfer of dissimilatory (bi)sulfite reductase revisited. J Bacteriol 187: 2203–2208
Acknowledgments
This project was supported by a grant from the Ramat-Hovav Industrial Council, Israel and the BMBF-MOST Cooperation in Water Technologies Grant WT-501. Special thanks are conveyed to the management and staff of the Ramat-Hovav Council for their cooperation. We thank Nachshon Siboni, Orr Shapiro, and Esti Kramarsky-Winter for technical support and useful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ben-Dov, E., Brenner, A. & Kushmaro, A. Quantification of Sulfate-reducing Bacteria in Industrial Wastewater, by Real-time Polymerase Chain Reaction (PCR) Using dsrA and apsA Genes. Microb Ecol 54, 439–451 (2007). https://doi.org/10.1007/s00248-007-9233-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-007-9233-2