
Abstract
In the present study, the species composition and potential metabolic activities of bacterial communities of reed Phragmites australis (Cav.) (Trin. ex Steudel) periphyton from Lake Velencei were studied by cultivation-based and metabolic fingerprinting methods. Serially diluted spring biofilm samples were used to test the community-level physiological profiling (CLPP) using BIOLOG microplates, and for plating onto different media. On the basis of their morphological, biochemical, and physiological test results, 173 strains were clustered by numerical analysis. Representatives of amplified ribosomal DNA restriction analysis (ARDRA) groups were identified by their 16S rDNA sequence comparison. Based on the results of the CLPP investigations, regional differences were detected among the utilized substrate numbers and types, parallel with the increase in incubation time. The phenotypic test results of the strains showed considerable variability with respect to the sampling sites and the media used for cultivation. The most frequently isolated strains were identified as members of genera Agrobacterium, Pseudomonas (P. anguilliseptica, P. marginalis, P. alcaligenes, P. fragi) with aerobic or facultative anaerobic respiratory metabolism, and the species Aeromonas sobria and A. veronii with strong facultative fermentative metabolism. Other strains were identified as Gram-positive Arthrobacter, Bacillus, and Kocuria species. The rarely isolated strains were members of β-Proteobacteria (Acidovorax, Delftia, Hydrogenophaga, and Rhodoferax), γ-Proteobacteria (Psychrobacter and Shewanella), low G + C Gram-positives (Brevibacillus, Paenibacillus, and Exiguobacterium) and high G + C Gram-positives (Aureobacterium and Microbacterium).
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Lake Velencei is a shallow soda lake in Hungary. The western part of the lake with extensive reed coverage is a nature conservation area with a unique wildlife, whereas the middle and eastern parts with larger open waters function as recreational areas. After the basin was reconstructed between 1962 and 1985, the structure of reed stands in this shallow, turbid, and alkaline lake has changed, and various symptoms of reed decline have appeared in the eastern part of the lake. Except for two small creeks, the lake has no significant natural water inflows. The main water source is rainfall and the mostly hypertrophic water of the reservoirs, which is filtered through the macrophyte-dominated western part of the lake [23]. In the natural self-purification processes of the lake, not only the reeds play an important role by their mechanical filtration and sedimentation, but also the capacity and activity of the enormous biofilm microbial communities developing on the submerged parts of reed stems. In the past years, detailed cultivation-based microbiological investigations were performed to reveal the potential metabolic activities and species composition of aerobic and anaerobic bacterial communities of the sediment as well as the rhizome-associated bacterial communities of healthy and declining reed stands in Lake Velencei. Studies on the sediment indicated the presence of spatially and temporally variable chemoorganotrophic bacterial communities with a broad spectrum of biochemical and physiological activities [4]. The healthy and declining reed stands could be distinguished on the basis of their cultivable bacterial community structure [16].
Nowadays, several wastewater treatment processes (e.g., different constructed wetland systems) use the abilities of complex and changing plant-associated microbial communities participating in the mineralization of organic materials, detoxification, removal of contaminants, etc. [19, 28]. Although different cultivation-based and cultivation-independent monitoring techniques are available to detect the changes in the species diversity and metabolic potential of microbial communities associated with different wastewater treatment plants [1, 8, 30], little attention has been paid to the periphytic bacterial metabolic activity and species composition of natural wetland systems. The detection of the species diversity of microbial communities and the recognition of the various processes taking place in natural wetland systems may provide useful information in achieving a higher degree of elimination and overall stability in the operation of constructed wetlands.
Therefore, the aim of the present work was to obtain an insight on the potential metabolic activities and species diversity of reed periphyton bacterial communities in a Phragmites-dominated Hungarian shallow soda lake by applying cultivation-based 16S rDNA identification and BIOLOG metabolic profiling methods.
Materials and Methods
Description of the Sampling Sites and Sample Preparation
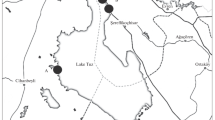
The surface of Lake Velencei (47°10′N, 18°35′E, Hungary) is about 25 km2, of which 9.8 km2 is covered by comparatively dense populations of common reed [Phragmites australis (Cav.) Trin ex Steudel]. The lake, which has an average depth of 1.4 m, features a sodium–magnesium–hydrocarbonate–sulfate dominance with 1000–3000 mg L–1 total dissolved salt and pH 7.8–10.0. For bacteriological investigations, 30-cm-long reed stem samples (in five replicates) were collected from 15 to 20 cm below the water surface at different locations [at Lángi-tisztás (LT) in the west, Gárdony (GT) in the middle, and Fürdető (FT) in the east] of the lake in April 2000 (Fig. 1). Physical and chemical parameters of the lake water at the time of sampling are presented in Table 1. Reed stem samples from the direction of the open water (external; K) as well as the closed and continuous stands (internal; B) were taken and immediately placed in known volume of sterile saline solution. Biofilm from the reed samples, following cooled transport to the laboratory, was washed with the help of a sterile brush.

Sampling sites of Lake Velencei. (LT: Lángi-tisztás; GT: Gárdony; FT: Fürdető; K: external reed stands; B: internal reed stands; gray area shows the reed coverage of the lake).
BIOLOG Carbon Source Utilization of Microbial Communities
Carbon source utilization patterns of reed biofilm communities were studied with the use of BIOLOG GN2 microplates. The wells of microplates containing 95 different carbon sources and a tetrazolium dye were inoculated with 150 μL diluted samples. The turbidity of samples was set according to the GN NENT standard provided by the manufacturer (Biolog Inc., Hayward, CA, USA). Development of the reduced purple color of the indicator due to substrate oxidation was measured at OD590 from 12 to 96 h of incubation at 25°C [25]. Absorbance data were examined with multivariate analysis [principal component analysis (PCA)] to determine the potential functional diversity of the microbial communities [20].
Isolation and Phenotypic Characterization of Bacterial Strains
Serially diluted reed biofilm samples were plated onto King’s (B) [6], tryptic soy yeast (TSY) DSMZ Medium 92 (T), Oligotrophic (O) [21] and Horikoshi-I alkaline DSMZ Medium 940 (A) media (http://www.dsmz.de/media/media.htm). After a 7- to 14-day incubation at 25°C, bacteria with different colony morphology were randomly isolated. The cell morphology, motility, Gram staining, and traditional biochemical–physiological tests (catalase and oxidase activities, reduction of nitrate, oxidation–fermentation catabolism of d-glucose, the methyl red and Voges–Proskauer reactions, aerobic acid production from d-glucose, hydrogen sulfide and indole production, hydrolysis of arginine, phenylalanine deamination, and hydrolysis of casein, gelatine, starch, and Tween 80) of the strains were investigated according to the procedure described by Smibert and Krieg [26]. Hierarchical clustering with UPGMA method and simple matching coefficient using the binary coded test results of the strains were performed by applying the SYN-TAX 2000 statistical software [20].
ARDRA, 16S rDNA Sequencing, and Phylogenetic Analysis
One loopful from colonies of the strains grown on agar surface for 24 h at 25°C were dispersed in 400 μL saline–EDTA buffer (150 mM NaCl, 10 mM EDTA; pH 8.0). A 5-μL lysozyme solution (10 mg mL−1) was added, and samples were incubated at 37°C for 30 min. Thereafter, 5 μL proteinase K (15 mg mL−1) and 10 μL 25% SDS were added, followed by incubation at 55°C for 30 min. DNA was extracted with equal volumes of phenol and chloroform [22]. After centrifugation, DNA was recovered from the aqueous phase using a PCR-M™ Clean-up System (Viogene, Sunnyvale, CA, USA). Polymerase chain reaction (PCR) amplification of 16S rDNA fragments was performed with 27f (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492r (5′-TACGGYTACCTTGTTACGACTT-3′) “universal” eubacterial primer pair in a Biometra T personal thermocycler. The PCR mixtures contained 1 U Taq DNA polymerase in the manufacturer’s buffer 1.5 mM MgCl2, 200 μM dNTPs and 0.3 μM of each primer (Fermentas, Hanover, MD, USA). Samples were first denatured at 98°C for 3 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s, and extension at 72°C for 1 min. Amplification was finished with a final extension step at 72°C for 7 min. PCR products were verified in 1% agarose gel.
ARDRA was carried out to obtain groups of strains with similar restriction patterns. Restriction digestion was performed with enzymes HinP1I and Tru9I, as described by Massol-Deya et al. [13]. The fragments were analyzed by agarose gel electrophoresis, and restriction patterns were visually compared. 16S rDNA-specific PCR products of the ARDRA group representatives were purified and concentrated by using the PCR-M™ Clean up System (Viogene). Sequencing reaction was accomplished by using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit v 3.1 (Applied Biosystems, Foster, CA, USA) with 519r (5′-GWATTACCGCGGCKGCTG-3′) primer according to the manufacturer’s protocol. Electrophoresis of the sequencing products was done in a Model 310 Genetic Analyser (Applied Biosystems). Phylogenetic analysis of the sequences was carried out by using the RDP II database and the ARB program [27]. Evolutionary distances were calculated via the algorithm of Kimura 2, and the phylogenetic tree was constructed with the neighbor-joining method.
Results and Discussion
The metabolic potential of reed biofilm microbial communities was estimated on the basis of substrate richness (number of positive wells, where the absorbance values were higher than the averages of absorbance values on a given microplate) [7, 9]. As Fig. 2 indicates, the average substrate richness of the five replicates increased in all samples originating from different sites of the lake during incubation time, and from the low initial values the ratio of the utilized carbon sources reached 74–84%. Differences among the average substrate richness of the different biofilm samples (originating either from external or internal reed stands) were the largest (29%) after 24 h of incubation, and decreased to 10% at 96 h of incubation. Among the studied sampling sites, differences in substrate richness were the smallest in the case of microbial communities from the western portion of the lake (Lángi-tisztás). In all microbial communities, the preferred carbon sources were polymers (88–100%) and carbohydrates (65–86%), whereas amines (22–46%) and carboxylic acids (14–38%) were the least utilized. PCA of the 96-h absorbance values of each well was also performed to describe the variability within reed stands. In the PCA, samples were examined as objects, and carbon sources as variables. According to the ordination, the first two PC axes accounted for 57% of the variance in the data (Fig. 3). In the ordination, samples originating from Fürdető (FTK and FTB) had the highest object scores for axis 1 (>1.5). Variability within the metabolic potential of the reed stem microbial communities from a given sampling site was also the smallest in the case of Fürdető. The differences among the reed biofilm samples from the different sampling sites (L, G, and F) were more definite than those among the sample types (K or B; external or internal reed stem samples).

Changes in average substrate richness of the reed (Phragmites australis) periphyton microbial communities on BIOLOG GN2 microplates after different incubation times originating from different sampling sites of Lake Velencei. (Data are mean values of the average of five replicates from a given sampling site).

Scores plot of principal component analysis based on 96-h absorbance data of BIOLOG GN2 microplates inoculated with reed (P. australis) periphyton samples from Lake Velencei. (Characters indicate the sampling sites and numbers the replicates of the reed stems).
More than 250 bacterial strains were isolated from the surface washings of reed stem biofilm from different parts of Lake Velencei. Most of the strains isolated from alkaline medium lost their viability; therefore, a total of 173 strains (56 from the Lángi-tisztás, 64 from the Agárd and 53 from the Fürdető samples) were thoroughly studied during the long-term laboratory investigations.
PCA was also used to evaluate the phenotypic variability of strain groups isolated from reed periphyton samples from Lake Velencei among different sampling sites and media used. In this ordination, bacterial strain groups (consisting of 13–26 strains isolated from the same sampling site and medium) were regarded as objects and the percentile positive results of the studied phenotypic characters of these strains as variables. The biplot in Fig. 4 simultaneously visualize both the strain groups from a given sampling site and medium (columns) and the phenotypic characters (rows) of the reduced data matrix. Therefore, in Fig. 4 bacterial strain group scores and phenotypic loadings (vectors) that describe the relative contributions of the characters in the ordination of the data matrix can be interpreted. In the biplot, all the strain groups from the Lángi-tisztás (LA, LB, and LT) and Gárdony (GA, GB, and GT) samples, with the exception of those isolated from oligotrophic medium (GO and LO), clearly separated from the ones of the Fürdetö samples (FB, FT, and FO). The strain groups cultivated from oligotrophic medium showed the least variability among the three sampling sites. Strain groups originating from the Fürdetö samples had the largest metabolic potential similar to the BIOLOG community-level physiological profiles. Strain groups from the alkaline medium (LA and GA) had the smallest metabolic potential in the studied biochemical tests, whereas those from King’s and TSY media (LB, GB, LT, and GT) showed the most similar metabolic potentials.

Two-dimensional principal components biplot describing phenotypic variability in bacterial strain groups isolated from reed (P. australis) periphyton samples from Lake Velencei among different sampling sites and media used. Sampling sites – L: Lángi-tisztás; G: Gárdony; F: Fürdetö. Media used – A: alkaline; B: King’s; T: TSY; O: oligotrophic.
Twenty-four clusters with two or more strains were established with at least 80% similarity level based on the hierarchical cluster analysis of the 27 binary coded test results of the 173 bacterial strains. Representatives with different ARDRA patterns (altogether 38 strains) were partially sequenced. The 16S rDNA-based phylogenetic analysis revealed that most of the strains (92) affiliated with γ-Proteobacteria. Almost identical numbers of strains were related to α-Proteobacteria (24), Gram-positive bacteria with high (24) and low (21) G + C DNA content. The remaining 12 strains were found to be members of β-Proteobacteria. The phylogenetic tree shown in Fig. 5 represents the phylotypes of ARDRA representatives obtained in this study and their closest relatives.

16S rDNA sequence based neighbor-joining phylogenetic tree of bacterial strains isolated from reed (P. australis) periphyton samples originating from Lake Velencei (Hungary) and selected reference sequences.
The three largest phenotypic clusters contained more than half (52%) of the strains. These strains stained Gram negatively and occurred at each sampling site of the lake. The 43 motile strains of the most abundant cluster showed a wide-ranging biochemical potential with catalase and oxidase positive reactions, reduction of nitrate to nitrite, a very intensive d-glucose oxidative and fermentative utilization with gas production, positive Voges–Proskauer and Simmons’ citrate reactions, production of indole from trytophane, and hydrolysis of arginine, aesculine, casein, gelatine, starch, and Tween 80 (Table 2). These strains were identified as Aeromonas sobria (Fig. 5). Other four strains with very similar metabolic profiles, additional positive hydrogen sulfide production, proved to be A. veronii (Fig. 5). The widely distributed species of the genus Aeromonas are typical members of aquatic habitats, exhibiting a marked seasonal cycle, and reach their highest abundance during the summer period [17]. Therefore, the appearance of these versatile strains in the reed biofilm at each sampling site may be a consequence of the increasing water temperature of Lake Velencei in April. In another Hungarian soda lake (Fertõ/Neusiedlersee), the dominant presence of A. sobria was also detected in the summer [11].
The altogether 41 strains from the second most abundant phenotypic cluster group were characterized by motility, positive catalase and oxidase reactions, hydrolysis of arginine, and Simmons’ citrate activity, and were determined as different species of the fluorescent group of Pseudomonas. Some characteristics, e.g., reduction of nitrate, ammonification, hydrogen sulfide production, hydrolysis of aesculin, casein, gelatine, and Tween 80 considerable varied among the strains of the clusters (Table 2). It is well known that most of the species of the genus Pseudomonas contain different plasmids and show significant metabolic flexibility, which allow them to decompose, among others, different toxic organic compounds (e.g., phenolics) [31]. Pseudomonas species are among the early colonizers of new natural habitats (e.g., plant surfaces) and frequently isolated members of soil, freshwater, and plant-associated microbial communities [12, 14, 32]. Most of our strains, originating mainly from the eastern Fürdető samples, were identified as P. anguilliseptica, generally found on fishes. Potential plant pathogenic (e.g., pectinolytic P. marginalis and P. fragi) and saproptrophic (P. putida) species were also detected among our isolates. Fluorescent pseudomonads, such as several strains of P. putida, with their siderophore (pyoverdine)-mediated iron-uptake system or their plant lectin-like bacteriocin (putidacin) production, are known as plant growth promoters [15, 18]. In contrast with aeromonads, Pseudomonas species often show psychrophilic or cold-tolerant properties, and are predominant members of the cultivable microbial communities of soils and fresh waters [3, 14].
Most of the 24 strains from the third largest cluster hydrolyzed arginine, phenylalanine, and aesculin, and yielded positive results in Simmons’ citrate and urease tests (Table 2). These strains showed 97–98% sequence similarity to Agrobacterium vitis (Fig. 5) and had strictly aerobic oxidative metabolism. The species comprised plant pathogenic (with Ti plasmids) or non-pathogenic strains, and are generally isolated from grapevine (Vitis spp.) and other (e.g., Actinidia spp.) dicotyledonous plant species [33]. The presence of Agrobacterium strains in association with a monocotyledonous plant, a declining reed (P. australis) rhizome, was reported by Micsinai et al. [16].
A large proportion of our strains (approximately 12%) belonged to Bacillus and closely related (Brevibacillus, Marinibacillus, and Paenibacillus) species of the low G + C division of Gram-positive bacteria. The low 16S rDNA sequence similarity values of our strains with Brevibacillus agri (96%) and a Paenibacillus sp. (95%) indicate that these strains may be isolates of new species. The Bacillus strains were generally active in the studied phenotypic tests but exhibited strain-dependent variable results (Table 2). Alkalitolerant (B. cereus and B. pumilus) and alkaliphilic (M. marinus) species may participate in the decomposition of different organic materials (e.g., plant polymers) by their wide-ranging metabolic potentials, and can tolerate the unfavorable conditions by forming endospores. The relative abundance of the facultative anaerobic chemoorganotrophic Bacillus and related strains is not surprising in shallow freshwater habitats, where they are frequently among the predominant species detected by cultivation [3, 4]. The primary habitats of these species can be the upper aerobic sediment regions, from where they probably colonize the submerged reed surfaces due to the strong wind-wave action. It may explain the fact that almost all of the strains were isolated from the central open water area (Gárdony) of the lake.
Strains belonging to the genera Kocuria, Micrococcus, and Arthrobacter among the high G + C division of Gram-positive bacteria originated from the western (Lángi-tisztás) and middle (Gárdony) parts of the lake (Table 2). It is interesting to note that the species Kocuria palustris was described from the rhizoplane of narrow-leaved cattail (Typha angustifolia) in a floating mat of the Soroksár Danube [10]. It is the second appearance of this species in a very similar habitat (western part of Lake Velencei), which is also dominated by floating reed mats. The aerobic respiratory species (K. rosea, Micrococcus luteus, and Arthrobacter crystallopoietes) are typical inhabitants of soils and aquatic environments with a broad-scale tolerance against environmental factors (e.g., salts, pH, and temperature).
Only a few strains identified as species Hydrogenophaga palleronii, Delftia acidovorans, and Acidovorax delafieldii represented β-Proteobacteria (Fig. 5). One of the most interesting common physiological properties of these strains is their ability to grow in aerobic, facultatively lithoautotrophic manner with hydrogen as electron donor. Acidovorax species were also demonstrated by fluorescent in situ hybridization (FISH) to be highly abundant in activated sludge samples of a municipal wastewater treatment plant [24], whereas the D24 strain of D. acidovorans, isolated from the Danube sediment north of Budapest, was able to mineralize atrazine (a widespread triazine herbicide) as the sole source of carbon and nitrogen [29].
Bacterial strains represented by only one or two species were also found within phylogenetic lineages of β-Proteobacteria (Rhodoferax fermentans), γ-Proteobacteria (Psychrobacter sp., Shewanella putrefaciens), high G + C Gram-positives (Aureobacterium kitamiense and Microbacterium imperiale), and low G + C Gram positives (Exigoubacterium sp.) (Fig. 5). These strains were isolated from the western (Lángi-tisztás) and middle (Gárdony) parts of Lake Velencei using King’s (B) and TSY media.
It is noteworthy that among the media used, TSY proved to be the most suitable for cultivation of different bacterial species (altogether 17) from the periphyton communities of reed (P. australis) samples. Strains of a Gram-negative species (Psychrobacter sp.), and four Gram-positive species (Paenibacillus sp., K. palustris, K. rosea, and Exiguobacterium sp.) were detected only on TSY medium. Those strains that were closely related to the Psychrobacter sp. and the Paenibacillus species, due to low (95–95%) 16S rRNA gene sequence similarity values and the discrepancies among the phenotypic properties, may represent new taxa. Conversely, Horikoshi-I alkaline medium proved to be the most selective one because only six different (mainly alkaliphilic or moderately halophilic) species (e.g., Marinibacillus marinus, Micrococcus luteus) were identified from it and, unfortunately, most of these strains lost their viability during the laboratory investigations.
Interestingly, when applying cultivation-based methods, almost identical species diversity was found in the reed periphyton samples from the transitional middle (Gárdony) and the floating mat macrophyte-dominated western (Lángi-tisztás) regions of the lake (with 18 and 16 different species). This phenomenon can be the consequence of the thick layers of extracellular polysaccharide containing microbial biofilms, which can develop on the submerged surfaces of the reeds and the higher concentrations of dissolved organic material that reach the western part of the lake through the inflow channels from the reservoirs. Species belonging to β-Proteobacteria were demonstrated only from these parts of the lake. In polluted rivers the dominant presence of frequently plasmid containing β-Proteobacteria was also described, which may be connected to their higher tolerance of pollutants and heavy metals [2, 5]. In contrast with the relatively high species diversity of the western and middle parts of the lake, not more than 10 species, affiliated mostly with the dominant cosmopolitan freshwater bacteria of γ- and α-Proteobacteria, were determined in the samples collected from the large open water region (Fürdető). This part of the lake can be characterized by higher alkalinity and salt concentrations, as well as stronger wind-wave actions, which continuously have strong mechanical damage on biofilms developing on the submerged parts of reeds.
In conclusion, detection of BIOLOG metabolic profiles of microbial communities and cultivation of bacteria with the use of numerous different media in combination with 16S rDNA-based phylogeny proved to be suitable approaches for the description of not only the potential metabolic ability of microorganisms but the cultivable diversity of reed biofilm communities. With respect to the phylogenetic analysis of the reed (P. australis) periphyton isolates, the majority of the strains belonged to well-known autochthonous freshwater bacterial taxa; however, some strains according to their low 16S rDNA sequence similarity may belong to new candidate species. The studied potential metabolic activities of the uncultured bacterial communities as well as the cultivated bacterial species indicate that the complex microbial communities of the reed periphyton may adapt to the varying parameters of the plant–microbe interactions. This is achieved by altering their metabolism from aerobic to anaerobic and/or from oxidative to fermentative and by showing wide-ranging decomposing or substrate utilizing activities and/or tolerance of natural environmental compounds.
References
Amann, R, Lemmer, H, Wagner, M (1998) Monitoring the community structure of wastewater treatment plants: a comparison of old and new techniques. FEMS Microb Ecol 25: 205–215
Böckelmann, U, Manz, W, Neu, TR, Szewzyk, U (2000) Characterization of the microbial community of lotic organic aggregates (‘river snow’) in the Elbe River of Germany by cultivation and molecular methods. FEMS Microbiol Ecol 33: 157–170
Borsodi, AK, Farkas, I, Kurdi, P (1998) Numerical analysis of planktonic and reed biofilm bacterial communities of Lake Fertő (Neusiedlersee, Hungary/Austria). Water Res 32: 1831–1840
Borsodi, AK, Vladár P, Cech G, Gedeon, G, Beszteri B, Micsinai A, Reskóné, MN, Márialigeti, K (2003) Bacterial activities in the sediment of Lake Velencei, Hungary. Hydrobiologia 506–509: 721–728
Brümmer, IHM, Felske, A, Wagner-Döbler, I (2003) Diversity and seasonal variability of β-proteobacteria in biofilms of polluted rivers: analysis by temperature gradient gel electrophoresis and cloning. Appl Environ Microbiol 69: 4463–4473
Cowan, ST, Steel, KJ (1974) Manual for the Identification of Medical Bacteria. University Press, Cambridge
Garland, JL (1997) Analysis and interpretation of community-level physiological profiles in microbial ecology. FEMS Microb Ecol 24: 289–300
Ibekwe, AM, Grieve, CM, Lyon, SR (2003) Characterization of microbial communities and composition in constructed dairy wetland wastewater effluent. Appl Environ Microbiol 69: 5060–5069
Konopka, A, Oliver, L, Turco, RF Jr (1998) The use of carbon substrate utilization patterns in environmental and ecological microbiology. Microb Ecol 35: 103–115
Kovács, G, Burghardt, J, Pradella S, Schumann, P, Stackebrandt, E, Márialigeti, K (1999) Kocuria palustris sp. nov. and Kocuria rhizophila sp. nov., isolated from the rhizoplane of the narrow-leaved cattail (Typha angustifolia). Int J Syst Bacteriol 49: 167–173
Langó, Zs, Borsodi, AK, Micsinai, A (2002) Comparative studies on Aeromonas strains isolated from lakes Balaton (Hungary) and Fertő/Neusiedlersee (Hungary). Acta Microbiol Immun Hung 49: 37–45
Makk, J, Ács, É, Márialigeti, K, Kovács, G (2003) Investigations on the Danube gravel-biofilm diatom-associated bacterial communities. Biologia 58: 729–742
Massol-Deya, AA, Odelson, DA, Hickey, RF, Tiedje, JM (1995) Bacterial community fingerprinting of amplified 16S and 16–23S ribosomal DNA sequences and restriction endonuclease analysis (ARDRA). In: Akkermans, ADL, van Elsas, JD, deBruijn, FJ (Eds.) Molecular Microbial Ecology Manual. Kluwer Academic Publishers, Dordrecht, pp 3.3.2:1–8
Meyer, AF, Lipson, DA, Martin, AP, Schadt, CW, Schmidt, SK (2004) Molecular and metabolic characterization of cold-tolerant alpine soil Pseudomonas sensu stricto. Appl Environ Microbiol 70: 483–489
Meyer, JM, Stintzi, A, Coulanges, V, Shivaji, S, Voss, JA, Taraz, K, Budzikiewicz, H (1998) Siderotyping of fluorescent pseudomonads: characterization of pyoverdines of Pseudomonas fluorescens and Pseudomonas putida strains from Antarctica. Microbiology 144: 3119–3126
Micsinai, A, Borsodi, AK, Csengeri, V, Horváth, A, Oravecz, O, Nikolausz, M, Reskóné, MN, Márialigeti, K (2003) Rhizome-associated bacterial communities of healthy and declining reed stands in Lake Velencei, Hungary. Hydrobiologia 506–509: 707–713
Monfort, P, Baleux, B (1990) Dynamics of Aeromonas hydrophila, Aeromonas sobria and Aeromonas caviae in a sewage treatment pond. Appl Environ Microbiol 56: 2007–2011
Parret, AHA, Schoofs, G, Proost, P, De Mot, R (2003) Plant lectin-like bacteriocin from a rhizosphere-colonizing Pseudomonas isolate. J Bacteriol 185: 897–908
Pinney, ML, Westerhoff, PK, Baker, L (2000) Transformations in dissolved organic carbon through constructed wetlands. Water Res 34: 1897–1911
Podani, J (2001) SYN-TAX 2000 Computer Programs for Data Analysis in Ecology and Systematics (Users’ Manual)
Poindexter, JS (1991) Dimorphic prosthecate bacteria: the genera Caulobacter, Asticcacaulis, Hyphomicrobium, Hyphomonas and Thiodendron. In: Balows, A, Trüper, HG, Dworkin, M, Harder, W, Schleifer, K (Eds.) The Prokaryotes. Springer-Verlag, New York, pp 2176–2196
Rainey, FA, Rainey, WN, Kroppenstedt, RM, Stackebrandt, E (1996) The genus Nocardiopsis represents a phylogenetically coherent taxon and a distinct actinomycete lineage: proposal of Nocardiopsaceae fam. nov. Int J Syst Bacteriol 46: 1088–1092
Reskóné, MN, Borsodi, AK (2003) Long-term investigations on the changes of the MPN values of bacterial communities participating in the sulphur cycle in Lake Velencei, Hungary. Hydrobiologia 506–509: 715–720
Schulze, R, Spring, S, Amann, R, Huber, I, Ludwig, W, Schleifer, KH, Kämpfer, P (1999) Genotypic diversity of Acidovorax strains isolated from activated sludge and description of Acidovorax defluvii sp. nov. Syst Appl Microbiol 22: 205–214
Smalla, K, Wachtendorf, U, Heuer, H, Liu, WT, Forney, L (1998) Analysis of BIOLOG GN substrate utilization patterns by microbial communities. Appl Environ Microbiol 64: 1220–1225
Smibert, RM, Krieg, NR (1994) Phenotypic characterization. In: Gerhardt, P, Murray, RGE, Wood, WA, Krieg, NR (Eds.) Methods for General and Molecular Bacteriology. American Society for Microbiology, Washington, DC, pp 603–711
Strunk, O, Ludwig, W (1995) ARB—a software for environmental sequence data. Department of Microbiology. Technical University of Munich, Germany
Vacca, G, Wand, H, Nikolausz, M, Kuschk, P, Kästner, M (2005) Effect of plants and filter materials on bacterial removal in pilot-scale constructed wetlands. Water Res 39: 1361–1373
Vargha, M, Takáts, Z, Márialigeti, K (2005) Degradation of atrazine in a laboratory scale model system with Danube river sediment. Water Res 39: 1560–1568
Wagner, M, Loy, A, Nogueira, R, Purkhold, U, Lee, N, Daims, H (2002) Microbial community composition and function in wastewater treatment plants. Antonie van Leeuwenhoek 81: 665–680
Wand, H, Laht, T, Peters, M, Becker, PM, Stottmeister, U, Heinaru, A (1997) Monitoring of biodegradative Pseudomonas putida strains in aquatic environments using molecular techniques. Microb Ecol 33: 124–133
Worm, J, Nybroe, O (2001) Input of protein to lake water microcosms affects expression of proteolytic enzymes and the dynamics of Pseudomonas spp. Appl Environ Microbiol 67: 4955–4962
Young, JM, Kuykendall, LD, Martínez-Romero, E, Kerr, A, Sawada, H (2001) A revision of Rhizobium Frank 1889, with an emended description of the genus, and the inclusion of all species of Agrobacterium Cohn 1942 and Allorhizobium undicola de Lajudie et al. 1998 as new combinations: Rhizobium radiobacter, R. rhizogenes, R. rubi, R. undicola and R. vitis. Int J Syst Evol Microbiol 51: 89–103
Acknowledgment
This research was supported by the Hungarian National Science Foundation (OTKA) Grants T032444 and T038021.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Borsodi, A.K., Rusznyák, A., Molnár, P. et al. Metabolic Activity and Phylogenetic Diversity of Reed (Phragmites australis) Periphyton Bacterial Communities in a Hungarian Shallow Soda Lake. Microb Ecol 53, 612–620 (2007). https://doi.org/10.1007/s00248-006-9133-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-006-9133-x