
Abstract
A quantitative real-time polymerase chain reaction (PCR) assay was developed for monitoring naphthalene degradation during bioremediation processes. The phylogenetic affiliations of known naphthalene-hydroxylating dioxygenase genes were determined to target functionally related bacteria, and degenerate primers were designed on the basis of the close relationships among dioxygenase genes identified from naphthalene-degrading Proteobacteria. Evaluation of the amplification specificity demonstrated that the developed real-time PCR assay represents a rapid, precise means for the group-specific enumeration of naphthalene-degrading bacteria. According to validation with bacterial pure cultures, the assay discriminated between the targeted group of naphthalene dioxygenase sequences and genes in other naphthalene or aromatic hydrocarbon-degrading bacterial strains. Specific amplification of gene fragments sharing a high sequence similarity with the genes included in the assay design was also observed in soil samples recovered from large-scale remediation processes. The target genes could be quantified reproducibly at over five orders of magnitude down to 3 × 102 gene copies. To investigate the suitability of the assay in monitoring naphthalene biodegradation, the assay was applied in enumerating the naphthalene dioxygenase genes in a soil slurry microcosm. The results were in good agreement with contaminant mineralization and dot blot quantification of nahAc gene copies. Furthermore, the real-time PCR assay was found to be more sensitive than hybridization-based analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In bioremediation studies, the quantitative analysis of functional genes has provided a valuable tool for studying the relationship between specific microbial populations and the performance of the degradation processes [30, 34]. In the environment, however, a wide range of bacteria participate in the degradation of organic contaminants. Because diverse bacteria are associated with different phases of pollutant degradation [19, 34, 43], better interpretation of the microbial community dynamics occurring during the progression of decontamination is important in the development of more efficient remediation processes. It is essential to not only obtain quantitative information about specific catabolic functions, but also to relate this information to the phylogeny and biology of the corresponding microbial populations. Instead of monitoring a single genotype, one should focus on the detection of a certain biological function performed by a specific group of organisms [8].
In the present study, our aim was to develop a rapid and quantitative assay for the enumeration of a specific group of bacteria possessing a similar degradation potential. Because the biological degradation of naphthalene has been well characterized [4], it was chosen as a model compound. Naphthalene, a toxic aromatic hydrocarbon composed of two benzene rings, is often found in sites contaminated by coal tar and crude oil. In aerobic naphthalene degradation, the aromatic ring is oxidized during the first step of the degradation pathway. A hydroxylated intermediate that can be further degraded is formed in this dioxygenase-catalyzed reaction [4]. According to reports, naphthalene degradative potential is widely distributed in the bacterial phylogeny, and there is a wide-ranging sequence diversity in naphthalene-hydroxylating dioxygenase genes in species of different evolutionary origin and with different substrate specificity [7, 9, 20–22, 29, 39]. Despite this genetic diversity, there is also considerable homology among naphthalene-hydroxylating dioxygenases, and certain amino acid residues at the active site of the enzyme are conserved [28]. This conservation enables the design of assays that target not only a wide range of naphthalene-degrading bacteria but also a specific group with the desired characteristics. In microbial ecology, assays targeting diverse genotypes have made it possible to investigate the genetic diversity of intrinsic or isolated naphthalene-degrading bacteria [5, 6, 50]. In bioremediation studies, on the other hand, the presence of more specific naphthalene-degrading genotypes has been shown to characterize the degradation potential of hydrocarbon-contaminated environments [11, 41, 44].
Recently, Baldwin et al. [2] and Dionisi et al. [5] reported the development of a real-time polymerase chain reaction (PCR) analysis for the enumeration of specific groups of naphthalene and other aromatic hydrocarbon-degrading organisms. The method, based on continuous measurement of the fluorescence signal during amplification, allows rapid and precise quantification of target genes [48]. In environmental analysis, real-time quantitative PCR assays have also been developed for the determination of genes involved in the metabolism of other environmental contaminants such as arsenate, carbazole, 2-chlorobenzoate, and toluene [3, 38, 42, 45]. According to the analysis of contaminated sediment and soil samples [15, 38], the method possesses good potential in the quantification of specific biological functions in the environmental samples. However, there are still relatively few published applications of real-time PCR in the monitoring of environmental microbial processes [42].
With the aim of investigating the applicability of real-time PCR analysis in the quantitative assessment of an environmentally relevant group of diverse genotypes, we developed a novel real-time PCR assay targeting a phylogenetically closely related group of naphthalene-degrading bacteria. To enable the detection of diverse gene sequences, degenerate primers were designed on the basis of gene sequences coding for the large subunit of naphthalene dioxygenase. To further enhance the enumeration of different target bacteria possessing similar naphthalene degradation characteristics, instead of a single genotype, we used the SYBR Green-based detection system. Because the molecule emits fluorescence on binding to any double-stranded DNA [47], and therefore requires no predetermination of target sequences, it provides superior means for the detection of diverse target genes in comparison to the Taqman probe approach that is often employed in environmental applications [3, 5].
To evaluate whether the developed PCR assay would be applicable in the analysis of heterogeneous and biologically diverse soil samples contaminated with organic hydrocarbons, the method was applied in the enumeration of target genes in field-scale remediation process samples and during naphthalene mineralization in a soil slurry. Although many studies have demonstrated the application of PCR-based techniques in the analysis of contaminated groundwater [15, 26, 46], the enumeration of PAH-degrading bacteria in soil samples by means of PCR has been limited [33, 41]. In this study, we show, by using dot blot hybridization as validation method and 14C-mineralization as a direct indicator of naphthalene biodegradation, that real-time PCR provides a specific tool also for the monitoring of biological process performed by a certain group of bacteria.
Methods
Bacterial Strains and Environmental Samples
Pseudomonas putida G7 (DSM 4476) carrying the naphthalene catabolic nah pathway was used as the positive control. P. putida (DSM 291), Bacillus subtilis (DSM 10), Escherichia coli (DSM 1576), xylene-degrading P. putida strain mt-2 (DSM 3931), and toluene-degrading P. putida F1 (DSM 6899) served as the negative control strains for amplification. Naphthalene-degrading P. putida OUS82, Ralstonia sp. U2 (NCIB 13827), and Burkholderia sp. RP007 (ICMP 13529) were used to determine the target range of the assay.
Environmental samples were taken as a composite sample from contaminated soil (Table 1). The EkoPAH sample was recovered from PAH-contaminated soil that was stored in a pile at the Ekokem hazardous waste treatment plant in Riihimäki, Finland [30]. The SoilTar and SoilOil samples were obtained from the Soilrem bioremediation treatment plant in Virkkala, Finland. The SoilTar sample was taken from a pile where soil contaminated by wood preservation activities was being treated by composting. The SoilOil sample originated from old gas station plants and was being treated by composting and vapor extraction. The PAH content of the soil samples was analyzed using acetone–hexane extraction as described in [18]. The dry weight was determined by drying at 105°C overnight.
Microcosm
Naphthalene biodegradation was studied in a soil slurry microcosm previously described in [30]. In that study, P. putida G7 inoculated and noninoculated soil slurry containing 10% of EkoPAH soil was amended with 30 mg L1 naphthalene and 180,000 dpm 14C-naphthalene. The DNA samples were amended with unlabeled naphthalene only. Four replicate mineralization samples and three replicate samples for DNA analysis were collected on each sampling time during a period of 7 days. Naphthalene degradation was monitored on the basis of the production of 14CO2 [30]. DNA was extracted as described in the following.
Phylogenetic Analysis and Primer Design
Phylogenetic analysis of genes coding for the large subunit of naphthalene-hydroxylating dioxygenase in naphthalene-degrading bacteria was performed with the ARB software [23]. Published full-length sequences obtained from GenBank were aligned using the Fast Aligner tool of ARB, and the resulting alignments were checked and corrected manually. Phylogenetic trees were constructed on the basis of sequence pair dissimilarities. Evolutionary distances were calculated using the model of Jukes and Cantor [16], and the phylogenetic relationships were estimated by neighbor-joining analysis. Bootstrap analysis was performed by resampling the data 1000 times. In addition to neighbor-joining analysis, maximum-likelihood and maximum-parsimony algorithms were used as alternative tree construction methods. The partial gene sequences obtained from environmental samples in this study were added to the neighbor-joining tree by the parsimony procedure.
Based on phylogenetic analysis, 15 gene sequences were included in the primer design (Table 2). To allow the amplification of divergent target genes, one nucleotide degeneracy was included in both nahAF 5′-CCCYGGCGACTATGT-3′ and nahARev 5′-TGCGTCCAACCMACG-3′ primers. In addition, there was one nucleotide mismatch in the nahARev primer to the pahA3, nahAc, and ndoC2 genes of P. putida OUS82 and two P. fluorescens strains that was considered acceptable because of the otherwise high sequence similarity. The primers amplify a 489-bp gene fragment at nucleotide positions 150–638 of nahAc gene of P. putida G7. The nahAF primer corresponds to the degenerate Ac149f primer previously described in [6].
DNA Isolation
Plasmid DNA was extracted by the alkaline lysis method according to Sambrook and Russel [35]. Total DNA was prepared using the bead-mill homogenization protocol described in [37]. Following homogenization, the total DNA was precipitated with isopropanol and extracted with phenol/chloroform/isoamyl alcohol. The quantity of DNA was determined spectrophotometrically using the Ultrospec 2100 pro spectrophotometer (Biochrom, UK).
Quantification of Naphthalene Dioxygenase Genes by Real-Time PCR
Real-time PCR analysis was performed using the SYBR Green-based detection system. Two microliters of LightCycler Fast Start DNA Master SYBR Green I master mix, 2 mM of MgCl2, 10 pmol of each primer, and 2.5 μL of DNA extract were added to a total reaction volume of 20 μL. All DNA samples were analyzed as 100-fold dilutions from the original DNA extracts. PCR amplification was carried out in the LightCycler device (Roche Diagnostics, Germany) using a temperature transition rate of 20°C s1. The thermal profile included an initial denaturation step at 95°C for 10 min, followed by 38 cycles of denaturation at 95°C for 1 s, annealing at 57°C for 5 s, and elongation at 72°C for 20 s. Fluorescence intensity was measured in the single acquisition mode after each elongation step at 72°C. For the subsequent melting curve analysis, the temperature was increased to 95°C, immediately dropped to 60°C for 30 s, and then increased again to 95°C using the temperature transition rate of 0.1°C s1. Fluorescence was measured continuously during heating. Following melting curve analysis, the PCR products were also visualized on 1.2% agarose gel.
The acquired fluorescence data were analyzed with the LightCycler Data Analysis software (Roche Diagnostics, Germany). The baseline was adjusted with the arithmetic baseline adjustment algorithm, and threshold cycle values (C t) were extracted with the second-derivative maximum method. Melting curves were calculated using the polynomial algorithm. Melting temperatures (T m) for each amplification product were determined using the Manual T m option of the software.
The standard curve for the determination of gene copy numbers was established by analyzing different amounts of unlinearized plasmid DNA isolated from P. putida G7 pure culture. The mass of target genes in plasmid DNA preparations was determined by dot blot hybridization, and molecular mass of the nahAc gene was used to convert the mass of nahAc to gene copy numbers. Three independent serial dilutions containing 3 × 102–3 × 106 gene copies of nahAc were then prepared in water solution, and all standard DNA samples were analyzed in one analysis run to create an external standard curve. To quantify the amount of target gene in environmental samples, three standards containing 3 × 106, 3 × 105, or 3 × 104 gene copies were included in each analysis run. The C t values from the three standard samples were then compared to the corresponding mean values in the external standard curve. The differences were averaged, and the mean value was added to the C t values obtained from the analysis. The PCR efficiency (E) was determined from the slope of the external calibration curve according to E = 101/slope [32]. Because the targeted gene sequences share high sequence similarity, the calculated PCR efficiency was considered representative also for the other target genes.
Quantification of Naphthalene Dioxygenase Genes by Dot Blot Hybridization
Quantification of nahAc gene copies in P. putida G7 plasmid DNA and total DNA preparations performed in this study was conducted according to the protocol described in [30]. Standard dilutions, control samples, and DNA extracts of interest were denatured with 0.5 M NaOH and vacuum blotted on Hybond-N nylon membrane (Amersham Biosciences, UK). The naphthalene dioxygenase gene probe was 32P-labeled and hybridized on the membrane. The quantification of naphthalene dioxygenase gene copies in soil slurry microcosm by dot blot hybridization was earlier reported in [30]. The same three replicate DNA samples were analyzed by quantitative real-time PCR in this study.
Denaturing Gradient Gel Electrophoresis
Environmental DNA extracts were reamplified with nahAFGC and nahARev primers. nahAFGC corresponds to the nahAF primer described above, except that a 40-bp-long GC clamp [27] is attached to the 5′-end of the primer (5′-CGCCCGCCGCGCCGGCGGGCGGGGCGGGGGCACGGGGGGCCCYGGCGACTATGT-3′). The 50-μL reaction mixture was composed of 1× Dynazyme™ II buffer (10 mM Tris–HCl, pH 8.8, 1.5 mM MgCl2, 50 mM KCl, and 1% Triton-X-100), 0.2 mM of each dNTP, 5 pmol of each primer, and 1 U of Dynazyme™ II DNA polymerase (Finnzymes, Finland). Amplification was carried out in Eppendorf MasterCycler (Eppendorf, Germany) using the following PCR program: initial denaturation at 94°C for 5 min with 30 cycles of 45 s at 94°C, 1 min at 57°C, 45 s at 72°C, and a 10-min final elongation at 72°C. PCR products were separated by denaturing gradient gel electrophoresis (DGGE) on 7% acrylamide gel containing a denaturing gradient from 45 to 54% (100% denaturant corresponds to 7 M urea and 40% formamide). The gel was run in Decode Universal Mutation Detection System (Bio-Rad, USA) in 0.5 × TAE (1 × TAE is 20 mM Tris–acetate, 0.5 mM EDTA, pH 8.0) at 75 V for 19 h at 60°C. After staining with SYBR Green I (BMA, Denmark), the bands were visualized with Gel Doc 2000 UV Transilluminator (Bio-Rad).
Cloning and Sequencing of Amplified Gene Fragments
Visible bands were excised from the DGGE gel and amplified with nahAFGC and nahARev primers as described above. Freshly made PCR products were purified with PCR Purification kit (Qiagen, Germany) and cloned using TOPO TA Cloning® kit Version R (Invitrogen, UK). The sequence of 10 positive clones was determined from each cloning reaction. Cloned gene fragments were amplified with nahAF and nahARev primers and sequenced with BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, California, USA) using the ABI Prism 310 Genetic analyzer (Applied Biosystems). The sequencing was performed in both directions to acquire the whole sequence of the fragment.
Statistical Analysis
Statistically significant differences between the samples were determined using Student's two-tailed t-test with unequal variance.
Results
Phylogeny of the Naphthalene-Hydroxylating Dioxygenases
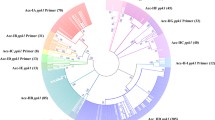
Phylogenetic analysis assigned the naphthalene-hydroxylating dioxygenase gene sequences from distinct bacterial isolates into three main clusters (clusters I, II, and III in Fig. 1). Cluster I contains naphthalene dioxygenase genes from gram-negative γ-Proteobacteria, sharing 87% or higher sequence similarity. The group also encompasses genes characterized from naphthalene-degrading Ralstonia sp. U2 and Comamonas testosteroni H with 78% sequence identity to Pseudomonas species. Cluster III consists of genes from gram-positive Rhodococcus species and Mycobacterium sp. PYR-1, sharing 59–91% sequence identity. According to neighbor-joining analysis, the third group, cluster II, includes the phnAc and bphA1f genes from gram-negative Burkholderia sp. RP007 and Sphingomonas aromaticivorans F199. All the inferred phylogenetic relationships were supported by bootstrap values ranging from 55 to 100% (Fig. 1). Moreover, maximum-likelihood and maximum-parsimony algorithms produced tree topologies similar to those obtained by neighbor-joining analysis. The only exception between the three algorithms was that the subgroup containing the genes from Burkholderia sp. RP007 and S. aromaticivorans F199 was assigned to the same cluster with other gram-negative species by the maximum-parsimony analysis.

A neighbor-joining tree showing the phylogenetic relationships among naphthalene-hydroxylating dioxygenase genes identified from different naphthalene-degrading bacteria. 2,3-Dihydroxybiphenyl 1,2-dioxygenase gene from Sphingomonas yanoikuyae B1 was used as an outgroup to root the tree. Numbers on the branches indicate percent bootstrap values with more than 50% bootstrap support. The scale bar represents 0.1 Jukes–Cantor substitutions per nucleotide. Partial sequences obtained from environmental samples are indicated with an asterisk.
Primer Design and Specificity of the Real-Time PCR Assay
Based on the inferred phylogenetic relationships and substrate specificity of the host bacteria, degenerate primers were designed for amplification of the conserved region in the large subunit of naphthalene dioxygenase genes assigned to cluster I and tested for their specificity. According to BLAST Search [1], the primer sequences were specific to naphthalene dioxygenase genes of the bacterial strains used for primer design. The primers were found to target also the ntdAc gene from 2-nitrotoluene degrading Pseudomonas sp. JS42 that shares 78% sequence similarity with the nahAc gene of P. putida G7. High sequence similarity to naphthalene dioxygenase genes from some uncultured bacteria sharing high sequence identity with the targeted gene sequences was also found, indicating that also other naphthalene catabolic genes closely related to the targeted genes could possibly be amplified with the designed primers.
The specificity of the developed assay and its capacity to distinguish between different naphthalene-degrading bacteria were also studied by determining the formation of amplification products in control samples (Fig. 2). Analysis of the bacterial strains used for primer design, P. putida G7, P. putida OUS82, and Ralstonia sp. U2, resulted in a gene fragment of the expected size. In contrast, no PCR product was formed from Burkholderia sp. strain RP007 that was not targeted by the assay. A weak fluorescence signal was detected during the late cycles of amplification in the P. putida (DSM 291) and E. coli negative control samples. In both samples, however, the signal was shown to be unspecific because of the abnormal melting behavior and incorrect length of the amplified fragments (Fig. 2). In addition, a weak unspecific amplification product was also observed in melting curve analysis and on agarose gel in the B. subtilis sample. However, no fluorescence signal could be measured during PCR amplification. In the environmental samples, positive control, and standard samples, the target gene was amplified specifically. A single melting peak was detected at 88.28°C (±0.12), and no primer dimers, or other unspecific products were formed. Thus, as the C t values measured in the negative control samples corresponded to the detection limit of the assay and were significantly higher than in any of the environmental DNA extracts analyzed, the analysis was considered accurate.

Specificity of the real-time PCR assay. Lane 1: Molecular weight marker; lane 2: nontemplate control; lane 3: B. subtilis total DNA; lane 4: E. coli total DNA; lane 5: P. putida (DSM 291) total DNA; lane 6: P. putida mt-2 total DNA; lane 7: P. putida F1 total DNA; lane 8: P. putida G7 total DNA; lane 9: P. putida OUS82 total DNA; lane 10: Ralstonia sp. U2 total DNA; lane 11: Burkholderia sp. RP007 total DNA; lanes 12–14: standards (3 × 104–3 × 106 gene copies); lane 15: EkoPAH soil sample; lane 16: SoilCreo soil sample; and lane 17: SoilOil soil sample.
Sequence Analysis of the Amplified Gene Fragments
The DGGE analysis of gene fragments amplified from environmental samples demonstrated that a single genotype was amplified in the EkoPah and SoilTar samples and two distinct gene fragments in the SoilOil sample (Fig. 3). All sequences showed 95–99% similarity at the DNA level. The NDOEkoPah and NDOSoilOil:1 clones were identical, whereas the NDOSoilTar clone amplified from tar-contaminated soil showed the greatest difference from the other sequences. According to the phylogenetic analysis, the NDOSoilOil:2 clone was identical with the nahAc gene of P. putida G7 (Fig. 1). The other clones were the most similar to naphthalene dioxygenase genes characterized from other Pseudomonas species, many of them corresponding to the genes that were used for primer design (Table 3; Fig. 1).

DGGE patterns of naphthalene dioxygenase gene fragments amplified from contaminated soil samples. Lane 1: Negative control; lane 2: P. putida G7 positive control; lanes 3 and 4: EkoPAH soil sample; lanes 5 and 6: SoilCreo soil sample; and lanes 7 and 8: SoilOil soil sample.
Quantification of the Gene Copy Numbers and the Detection Sensitivity of the Real-Time PCR Assay
Three replicate serial dilutions of plasmid DNA extracted from P. putida G7 were prepared to create a standard curve (Fig. 4). The relationship between the threshold values and log amount of target DNA was linear over five orders of magnitude (r 2 = 0.99), ranging from 3 × 102 gene copies to 3 × 106 gene copies of target gene. On this range of concentrations, the PCR efficiency was 1.84. When 3 × 100 and 3 × 101 gene copies of naphthalene dioxygenase were used for amplification, the linearity was lost and no PCR product was observed during melting curve analysis or on agarose gel. Therefore, the detection limit of the assay was determined to be 3 × 102 gene copies.

Sensitivity and dynamic range of the real-time PCR assay. The standard curve was generated by plotting the log amount of nahAc gene copies against the threshold values (C t) obtained from the analysis of three independent dilution series of standard DNA.
The Reproducibility and Accuracy of the Analysis
The intra-assay variability was evaluated using three replicate dilution series of standard DNA. The reproducibility between these three independent standard dilutions was good, although the deviation was, to some extent, dependent on the amount of target DNA. When 3 × 106 gene copies of the target gene were used, the variation in the threshold values was 1.5%, in contrast to variation of 2.4% for 3 × 102 gene copies. The results obtained from three separate analysis runs were also highly reproducible. Coefficients of variation calculated from standards containing 3 × 104, 3 × 105, and 3 × 106 gene copies were 2.18, 1.84, and 0.44%, respectively (n = 3). The accuracy of the analysis was evaluated by comparing the real-time PCR and dot blot hybridization results obtained from the analysis of P. putida G7 total DNA. When 1.9 × 108 gene copies of target gene in 1 μg of P. putida G7 total DNA were detected in real-time PCR analysis, the amount of target gene detected using dot blot hybridization was 1.3 × 108 gene copies.
Quantification of Naphthalene Dioxygenase Genes during Naphthalene Degradation Process
To further validate the developed assay, the real-time PCR results obtained during naphthalene degradation in the soil slurry microcosm were compared to the contaminant degradation and the dot blot hybridization results described in [30]. In the noninoculated soil slurry and in the P. putida G7 inoculated soil slurry, 2.2 × 106 and 9.6 × 107 gene copies in 1 g of soil dry weight (dw), respectively, could be detected by real-time PCR in the beginning of the study, whereas no target gene was observed by dot blot hybridization [30]. In the P. putida G7 inoculated soil slurry, naphthalene degradation was activated in the beginning of the study, and in 8 h time, the amount of target gene increased to a maximum of 2.3 × 109 gene copies in 1 g of soil (dw). In the noninoculated soil slurry, active mineralization was observed only after 28 h when the amount of target gene detected by PCR had reached the maximum number of 2.4 × 109 gene copies (Fig. 5). When the production of 14CO2 started to decline, there was a marked decrease in the amount of target gene, and 2.1 × 108 and 7.7 × 108 gene copies of target gene were detected with the PCR assay in the end of the study.

The amount of naphthalene dioxygenase genes in noninoculated soil slurry microcosm as determined by (a) real-time PCR (▴) and (b) dot blot hybridization (▵). The amount of naphthalene dioxygenase gene copies P. putida G7-inoculated soil slurry microcosm as determined by (c) real-time PCR (♦) and (d) dot blot hybridization (⋄) in noninoculated soil slurry. Naphthalene mineralization (•) was measured on the basis of 14CO2 production [30].
However, in both the noninoculated soil slurry and the P. putida G7 inoculated soil slurry, the PCR results differed from the amount of target gene detected by dot blot hybridization during the first 45 and 25 h, respectively. According to the real-time PCR analysis (Fig. 5a and c), the increase in the amount of target gene occurred earlier with respect to naphthalene mineralization than that detected by dot blot hybridization (Fig. 5b and d). Additionally, one order of magnitude difference was observed between the amount of target gene copies detected by the real-time PCR assay and dot blot hybridization in [30]. In the P. putida G7 inoculated soil slurry, the amount of target genes determined by PCR analysis reached the highest value after 8 h and decreased to 7.0 × 108 gene copies in 1 g of soil (dw) by 25 h, whereas according to dot blot analysis, the amount of target gene increased up until 25 h when 4.6 × 1010 copies were detected. In the noninoculated soil slurry, the amount of target gene copies determined by hybridization increased until 45 h reaching a maximum of 3.1 × 109 gene copies. In contrast, after 45 h, the amount of target genes detected by PCR analysis had already decreased to 2.7 × 109 gene copies. By the end of the study, the differences had balanced out in both treatments.
Quantification of Naphthalene Dioxygenase Genes in Field-Scale Remediation Process Samples
The target gene was successfully detected and quantified in all three hydrocarbon-contaminated soil samples. The highest amount of target gene, 4.5 × 106 gene copies per gram of soil (dw), was measured in the SoilTar sample containing the highest amount of total PAHs (Table 1). In the EkoPAH sample, 2.2 × 106 copies of target gene were detected in 1 g of soil (dw). The lowest amount of target gene, 1.2 × 106 gene copies, was measured in SoilOil sample contaminated with diesel and light fuel oil.
Discussion
In the present study, we applied real-time PCR analysis for studying a defined group of naphthalene-degrading bacteria in a contaminated environment and evaluated its applicability in monitoring remediation processes. During bioremediation, diverse bacteria possessing different degradation characteristics participate in pollutant degradation [8]. More detailed knowledge on the phylogenetic affiliation and catabolic potential of these organisms is needed to better understand the microbial ecology of contaminant degradation and, consequently, to estimate the efficiency of remediation processes [43]. To target a specific catabolic function, the naphthalene dioxygenase gene coding for the key step in aerobic naphthalene degradation was used as an indicator of naphthalene catabolic potential. As previous findings indicate, a relationship exists between the abundance of naphthalene dioxygenase gene and naphthalene degradation potential [41] and active contaminant degradation [30].
Because the genetic diversity among naphthalene-degrading bacteria is well demonstrated in the literature [7, 9, 20–22, 29, 39], phylogenetic analysis of naphthalene dioxygenase genes in distinct naphthalene-degrading bacterial isolates was performed to determine sequence similarities that would enable the detection of a defined group of bacteria. In agreement with the current view on the genetics of contaminant degradation, separate clusters of gene sequences could be designated on the basis of sequence pair dissimilarities and phylogenetic affiliation of the host bacterium [12]. However, the dioxygenase genes from Burkholderia sp. RP007 and S. aromaticivorans F199 could not be assigned with a high degree of confidence. This inconsistent clustering is most probably caused by the low sequence similarity to other known naphthalene-hydroxylating dioxygenases [22, 29].
Based on phylogenetic analysis, the cluster containing low molecular weight PAH-degrading bacteria belonging to γ- and β-subclass of Proteobacteria was chosen as a target group for primer design. By selecting this group of dioxygenase genes that were clearly separated from genes present in other naphthalene-degrading bacteria, the assay could be targeted at bacteria that possess specific degradation potential. Moreover, Proteobacteria species are known to be present in a number of contaminated environments and have been shown to become enriched after pollution [5, 25, 36], as well as to dominate the microbial community during the rapid pollutant degradation phase [17].
When broad target range PCR assays are designed for environmental applications, it is often not possible to ensure the specific amplification of target genes [40]. Demonstrating that the targeted real-time PCR assay developed herein was specific to the defined group of naphthalene-degrading bacteria, the designed primers did not amplify naphthalene-hydroxylating dioxygenase genes not targeted by the assay or other genes responsible for the hydroxylation of monoaromatic hydrocarbons. As shown by the specific amplification of the target genes in the environmental samples, the real-time PCR quantification of functional marker genes was suitable for studying specific microbial populations also in different environments. All the gene sequences amplified from contaminated soil samples were very similar, indicating a high functional similarity among the detected catabolic genes [28]. Especially in the soil contaminated with oil, the detection of two closely similar gene fragments clearly illustrated the feasibility of the real-time PCR quantification based on SYBR Green and degenerate primers in the assessment of degradative function instead of a single genotype. However, the identification of the amplified gene fragments was based on DGGE analysis. Because the method is known to detect species present only at higher cell densities [14], it is possible that some genotypes amplified with the degenerate primers may not have been detected in environmental samples. Wilson et al. [50] analyzed the sequence diversity of naphthalene dioxygenase transcripts amplified from coal tar-contaminated groundwater. According to their results, the dominating genotypes detected were similar to Pseudomonas-like dioxygenase genes, whereas bacteria showing higher sequence dissimilarity displayed lower gene expression. As our results demonstrate, all of the gene fragments were also closely related to genes used for primer design, supporting our aim of targeting a specific naphthalene catabolic activity in a group of phylogenetically closely related naphthalene-degrading bacteria.
The amount of target gene could be quantified on over five orders of magnitude, which is in good agreement with previous reports [2, 31]. According to the PCR efficiency, almost every PCR product was amplified during each cycle [32]. The results obtained by the method were highly reproducible, although the variation in the signals, as also pointed out by Grüntzig et al. [10], increased close to the detection limit. Also, the sensitivity of the real-time PCR assay was in good agreement with the detection limits in previously published PCR-based environmental studies [2, 13, 31].
To thoroughly evaluate the specificity and accuracy of the developed real-time PCR method in environmental analysis, the assay was applied in the enumeration of naphthalene dioxygenase genes in contaminated soil samples and naphthalene amended microcosm. In a laboratory-scale soil slurry microcosm, the changes in the amount of target gene determined by real-time PCR were highly consistent with contaminant degradation. In agreement with Wang et al. [42] and our previous results on hybridization analysis of nahAc genes in soil slurry microcosm [30], the number of gene copies increased during the course of naphthalene mineralization, and a decrease in the number of gene copies was detected after a decline in the production of 14CO2.
However, during active naphthalene mineralization, there were significant differences in the growth rate of naphthalene-degrading bacteria between PCR analysis and dot blot quantification performed previously [30] (Fig. 5). This is in contrast to Beller et al. [3] who demonstrated good agreement between the results from slot blot hybridization and real-time PCR during anaerobic toluene degradation. Because PCR-based techniques are sensitive to impurities and heterogeneous nontarget DNA commonly present in environmental samples [49], it is possible that in soil samples impurities may have inhibited the efficient amplification of target sequences. However, because a 10-fold difference was observed between the amount of target genes determined by real-time PCR method and hybridization analysis in our previous study [30], it is more likely that the observed discrepancies are caused by differences in the detection limits and dynamic ranges of the respective assays. As the detection of the target gene in the beginning of the biodegradation study by real-time PCR but not by dot blot hybridization demonstrates, the real-time PCR method was more sensitive than the hybridization-based technique. According to the analysis of fecal samples [24], the sensitivity of real-time PCR can be several times higher than that of dot blot hybridization. In addition, because the results from the quantification of nahAc gene copies in P. putida G7 total DNA in this study were highly consistent, it is likely that the one-order-of-magnitude difference observed between the two studies is a result of the dissimilarities in standard curve generation. As the gene copy numbers determined by real-time PCR were in good agreement with contaminant mineralization, a direct indicator of biodegradation, the results show that the developed method is suitable for accurate monitoring of microbial communities during contaminant degradation. This is in agreement with other studies, where catabolic activities have been compared to decrease in contaminant concentration [15, 46]. However, in these studies, direct comparison between the actual biotransformation rate and gene copy numbers cannot be determined.
According to the analysis of PAH-, tar-, and oil-contaminated soil samples, the developed assay is also applicable in the enumeration of naphthalene-degrading bacteria in large-scale remediation processes. In the analyzed samples, the highest amount of target gene was detected in tar-contaminated soil containing the highest concentration of aromatics. This is in good agreement with previous studies in which the amount of naphthalene dioxygenase genes identified from Proteobacteria have been shown to correlate with the contaminant content in polluted environment [5, 25].
In conclusion, our results illustrate that, with careful primer design, PCR assays can provide a powerful tool for studying biological functions in the environment. By using naphthalene dioxygenase genes from species in the γ- and β-subclass of Proteobacteria as a model group, we developed an assay targeting a phylogenetically closely related group of bacteria with similar naphthalene degradation potential. With the assay, we were able to acquire quantitative information on the distribution and growth of this group of functionally related bacteria in environmental samples. Moreover, it was shown how this quantitative information obtained from real-time PCR analysis can be linked to contaminant degradation and the progress of the remediation process. As more information on the degradation of other contaminants becomes available, the approach described herein will enable the design of more primer sets corresponding to other bacterial groups with specific degradation characteristics and evolutionary origin. As a result, the reliability of environmental process monitoring by molecular biological methods is increased.
References
Altschul, SF, Madden, TL, Schaffer, AA, Zhang, J, Zhang, Z, Miller, W, Lipman, DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402
Baldwin, BR, Nakatsu, CH, Nies, L (2003) Detection and enumeration of aromatic oxygenase genes by multiplex and real-time PCR. Appl Environ Microbiol 69: 3350–3358
Beller, HR, Kane, SR, Legler, TC, Alvarez, PAA (2002) A real-time polymerase chain reaction method for monitoring anaerobic, hydrocarbon-degrading bacteria based on a catabolic gene. Environ Sci Technol 36: 3977–3984
Cerniglia, CE (1984) Microbial metabolism of polycyclic aromatic hydrocarbons. Adv Appl Microbiol 30: 31–71
Dionisi, HM, Chewning, CS, Morgan, KH, Menn, FM, Easter, JP, Sayler, GS (2004) Abundance of dioxygenase genes similar to Ralstonia sp. strain U2 nagAc is correlated with naphthalene concentrations in coal tar-contaminated freshwater sediments. Appl Environ Microbiol 70: 3988–3995
Ferrero, M, Llobet-Brossa, E, Lalucat, J, García-Valdés, E, Rosselló-Mora, R, Bosch, R (2002) Coexistence of two distinct copies of naphthalene degradation genes in Pseudomonas strains isolated from the western Mediterranean region. Appl Environ Microbiol 68: 957–962
Fuenmayor, SL, Wild, M, Boyes, AL, Williams, PA (1998) A gene cluster encoding steps in conversion of naphthalene to gentisate in Pseudomonas sp. strain U2. J Bacteriol 180: 2522–2530
Futamata, H, Haryama, S, Watanabe, K (2001) Group-specific monitoring of phenol-hydroxylase genes for a functional assessment of phenol-simulated trichloroethylene bioremediation. Appl Environ Microbiol 67: 4671–4677
Goyal, AK, Zylstra, GJ (1997) Genetics of naphthalene and phenanthrene degradation by Comamonas testosteroni. J Ind Microbiol Biotech 19: 401–407
Grüntzig, V, Nold, SC, Zhou, J, Tiedje, JM (2001) Pseudomonas stutzeri nitrite reductase gene abundance in environmental samples measured by real-time PCR. Appl Environ Microbiol 67: 760–768
Guo, C, Sun, W, Harsh, JB, Ogram, A (1997) Hybridization analysis of microbial DNA from fuel oil-contaminated and noncontaminated soil. Microb Ecol 34: 178–187
Habe, H, Omori, T (2003) Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem 67: 225–243
Henry, S, Baudoin, E, Lopez-Gutierrez, JC, Martin-Laurent, F, Brauman, A, Philippot L (2004) Quantification of denitrifying bacteria in soils by nirK gene targeted real-time PCR. J Microbiol Methods 59: 327–335
Heuer, H, Wieland, G, Schönfeld, J, Schönwälder, A, Gomes, NCM, Smalla, K (2001) Bacterial community profiling using DGGE or TGGE analysis. In: Rochelle, PA (Ed.) Environmental Molecular Microbiology: Protocols and Applications. Horizon Scientific Press, UK, pp 177–190
Hosoda, A, Kasai, Y, Hamamura, N, Takahata, Y, Watanabe, K (2005) Development of a PCR method for the detection and quantification of benzoyl-CoA reductase genes and its application to monitored natural attenuation. Biodegradation 16: 591–601
Jukes, TH, Cantor, CR (1969) Evolution of protein molecules. In: Munro, HN (Ed.) Mammalian Protein Metabolism, vol. 3, Academic Press, New York, pp 21–132
Kaplan, CW, Kitts, CL (2004) Bacterial succession in a petroleum land treatment unit. Appl Environ Microbiol 70: 1777–1786
Karstensen, KH (1996) Nordic guideline for chemical analysis of contaminated soil samples. Nordtest, Espoo, NT Techn Report 329
Katsivela, E, Moore, ERB, Kalogerakis, N (2004) Monitoring of the microbial activities and the diversity of the microbial community degrading refinery waste sludge. Water Air Soil Pollut Focus 4: 75–85
Khan, AA, Wang, RF, Cao, WW, Doerge, DR, Wennerstrom, D, Cerniglia, CE (2001) Molecular cloning, nucleotide sequence, and expression of genes encoding a polycyclic aromatic ring dioxygenase from Mycobacterium sp. strain PYR-1. Appl Environ Microbiol 67: 3577–3585
Larkin, MJ, Allen, CC, Kulakov, LA, Lipscomb, DA (1999) Purification and characterization of a novel naphthalene dioxygenase from Rhodococcus sp. strain NCIMB12038. J Bacteriol 181: 6200–6204
Laurie, AD, Llouyd-Jones, G (1999) The phn genes of Burkholderia sp. RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J Bacteriol 181: 531–540
Ludwig, W, Strunk O, Westram, R, Richter, L, Meier, H, Yadhukumar, Buchner A, Lai T, Steppi, S, Jobb, G, Förster, W, Brettske, I, Gerber, S, Ginhart, AW, Gross, O, Grumann, S, Hermann, S, Jost, R, König, A, Liss, T, Lüßmann, R, May M, Nonhoff, B, Reichel, B, Strehlow, R, Stamatakis, A, Stuckmann, N, Vilbig, A, Lenke M, Ludwig, T, Bode, A, Schleifer, KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371
Malinen, E, Kassinen, A, Rinttila, T, Palva, A (2003) Comparison of real-time PCR with SYBR Green I or 5′-nuclease assays and dot-blot hybridization with rDNA-targeted oligonucleotide probes in quantification of selected faecal bacteria. Microbiology 149: 269–277
Margesin, R, Labbe, D, Schinner, F, Greer, CW, Whyte, LG (2003) Characterization of hydrocarbon-degrading microbial populations in contaminated and pristine Alpine soils. Appl Environ Microbiol 69: 3085–3092
Mesarch, MB, Nakatsu, CH, Nies, L (2004) Bench-scale and field-scale evaluation of catechol 2,3-dioxygenase specific primers for monitoring BTX bioremediation. Water Res 38: 1281–1288
Muyzer, G, de Waal, EC, Uitterlinden, AG (1993) Profiling complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59: 695–700
Parales, RE (2003) The role of active site residues in naphthalene dioxygenase. J Ind Microbiol Biotech 30: 271–278
Pinyakong, O, Habe, H, Omori, T (2003) The unique aromatic catabolic genes in sphingomonads degrading polycyclic aromatic hydrocarbons (PAHs). J Gen Appl Microbiol 49: 1–19
Piskonen, R, Nyyssönen, M, Rajamäki, T, Itävaara, M (2005) Monitoring of accelerated naphthalene-biodegradation in a bioaugmented soil slurry. Biodegradation 16: 127–134
Qiu, XY, Hurt, RA, Wu, LY, Chen, CH, Tiedje, JM, Zhou, JZ (2004) Detection and quantification of copper-denitrifying bacteria by quantitative competitive PCR. J Microbiol Methods 59: 199–210
Rasmussen, S (2001) Quantification on the LightCycler. In: Meuer, S, Wittwer, C, Nagakawara, K, (Eds.) Rapid Cycle Real-Time PCR, Methods and Applications. Springer Press, Heidelberg, pp 21–34
Rhee, SK, Liu, X, Wu, L, Chong, SC, Wan, X, Zhou, J (2004) Detection of genes involved in biodegradation and biotransformation in microbial communities by using 50-mer oligonucleotide microarrays. Appl Environ Microbiol 70: 4303–4317
Ringelberg, DB, Talley, JW, Perkins, EJ, Tucker, SG, Luthy, RG, Bouwer, EJ, Fredrickson, HL (2001) Succession of phenotypic, genotypic, and metabolic community characteristics during in vitro bioslurry treatment of polycyclic aromatic hydrocarbon-contaminated sediments. Appl Environ Microbiol 67: 1542–1550
Sambrook, J, Russel, DW (2001) Molecular Cloning, A Laboratory Manual, Ed. 3. Cold Spring Harbor Press, Cold Spring Harbour, NY
Shen, Y, Stehmeier, LG, Voordouw, G (1998) Identification of hydrocarbon-degrading bacteria in soil by reverse sample genome probing. Appl Environ Microbiol 64: 637–645
Stephen, JR, Chang, YJ, Gan, YD, Peacock, A, Peacock, SM, Pfiffner, SM, Barlecona, MJ, White, DC, Macnaughton, SJ (1999) Microbial characterization of a JP-4 fuel contaminated site using a combined lipid biomarker/PCR-DGGE based approach. Environ Microbiol 1: 231–241
Sun, Y, Polishchuk, EA, Radoja, U, Cullen, WR (2004) Identification and quantification of arsC genes in environmental samples by using real-time PCR. J Microbiol Methods 58: 335–349
Takizawa, N, Kaida, N, Torigoe, S, Moritani, T, Sawada, T, Satoh, S, Kiyohara, H (1994) Identification and characterization of genes encoding polycyclic aromatic hydrocarbon dioxygenase and polycyclic aromatic hydrocarbon dihydrodiol dehydrogenase in Pseudomonas putida OUS82. J Bacteriol 176: 2444–2449
Taylor, PM, Medd, JM, Schoenborn, L, Hodgson, B, Janssen, PH (2002) Detection of known and novel genes encoding aromatic ring-hydroxylating dioxygenases in soils and in aromatic hydrocarbon-degrading bacteria. FEMS Microbiol Lett 216: 61–66
Tuomi, M, Salminen, JM, Jorgensen, KS (2004) The abundance of nahAc genes correlates with the 14C-naphthalene mineralization potential in petroleum hydrocarbon-contaminated oxic soil layers. FEMS Microbiol Ecol 51: 99–107
Wang, G, Gentry, TJ, Grass, G, Josephson, K, Rensing, C, Pepper, IL (2004) Real-time PCR quantification of a green fluorescent protein-labelled, genetically engineered Pseudomonas putida strain during 2-chlorobenzoate degradation in soil. FEMS Microbiol Lett 233: 307–314
Watanabe, K, Futamata, H, Harayama, S (2002) Understanding the diversity in catabolic potential of microorganisms for the development of bioremediation strategies. Antonie Van Leeuwenhoek 81: 655–663
Whyte, LG, Bourbonnière, LG, Bellerose, CG, Greer, CW (1999) Bioremediation assessment of hydrocarbon-contaminated sediments from the high arctic. Bioremediat J 3: 69–80
Widada, J, Noriji, H, Kasuga, K, Yoshida, T, Habe, H, Omori, T (2001) Quantification of the carbazole 1,9a-dioxygenase gene by real-time competitive PCR combined with co-extraction of internal standards. FEMS Microbiol Lett 202: 51–57
Wikstrom, P, Hagglund, L, Forsman, M (2000) Structure of a natural microbial community in a nitroaromatic contaminated groundwater is altered during biodegradation of extrinsic, but not intrinsic substrates. Microb Ecol 39: 203–210
Witzthum, F, Geiger, G, Bisswanger, H, Brunner, H, Bernhagen, J (1999) A quantitative fluorescence-based microplate assay for the determination of double-stranded DNA using SYBR Green I and a standard ultraviolet transilluminator gel imaging system. Anal Biochem 276: 59–64
Wilhelm, J, Pingoud, A (2003) Real-time polymerase chain reaction. ChemBioChem 4: 1120–1128
Wilson, IG (1997) Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 63: 3741–3751
Wilson, MS, Bakermans, C, Madsen, EL (1999) In situ, real-time catabolic gene expression: extraction and characterization of naphthalene dioxygenase mRNA transcripts from groundwater. Appl Environ Microbiol 65: 80–87
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nyyssönen, M., Piskonen, R. & Itävaara, M. A Targeted Real-Time PCR Assay for Studying Naphthalene Degradation in the Environment. Microb Ecol 52, 533–543 (2006). https://doi.org/10.1007/s00248-006-9082-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-006-9082-4