
Abstract
A new method based on quantitative real-time polymerase chain reaction (qPCR) was developed and applied to quantify the red tide dinoflagellate Lingulodinium polyedrum in natural seawater samples and in laboratory cultures. The method uses a Molecular Beacon™ approach to target a species-specific region of the small subunit ribosomal RNA gene. The accuracy of the method was verified by microscopical counts using cultures of the dinoflagellate isolated from coastal waters near Los Angeles, CA, and with natural water samples spiked with cultured L. polyedrum. The method was applied to document the pattern and timing of vertical migration by the dinoflagellate in a 2-m water column on an 11:13 h light/dark photoperiod established in the laboratory. Positive phototaxis of L. polyedrum resulted in dense aggregations of the dinoflagellate within the top few centimeters of the water column during the light period. This pattern of distribution was readily established by both methods, although abundances of L. polyedrum determined using qPCR were higher than abundances determined by microscopy in the morning and lower in the afternoon and evening. These differences may have been a consequence of variability in the DNA content per cell because of synchrony of cell division. Counts using both methods to analyze natural samples collected from coastal waters in the Long Beach–Los Angeles area and adjacent San Pedro Channel were in close agreement. However, the qPCR method exhibited greater sensitivity than the microscopical method when L. polyedrum was present at low abundances, and qPCR had a much higher rate of sample throughput than microscopy. The development of this new approach for enumerating L. polyedrum provides a useful tool for studying the ecology of this important red tide species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Harmful algal blooms (HABs) appear to be increasing in frequency in many coastal environments worldwide [4, 28, 66, 67]. Many blooms of dinoflagellates (often referred to as “red tides”) form HABs that are toxic to other biota and can cause significant economic losses [34]. Little is known regarding the relative importance of the key factors governing these blooms because of the complex interplay between the physical, chemical, and biological factors involved [4]. Factors that appear to contribute to the formation of harmful dinoflagellate blooms include nutrient loading in coastal waters, advection and aggregation of populations, and introduction of species via ballast water discharge [5, 6, 29, 58, 65].
Lingulodinium polyedrum (Stein) Dodge (Gonyaulax polyedra) is a marine photosynthetic dinoflagellate that is often responsible for red tides. This bioluminescent species is widely distributed in warm temperate and subtropical waters of coastal areas [17, 44, 68] and has been associated with fish and shellfish mortality events [44, 73]. The presence of yessotoxin, a toxin regarded as hepatotoxic and cardiotoxic, was demonstrated in phytoplankton including L. polyedrum in the Adriatic Sea [18, 74]. Yessotoxin production by L. polyedrum was confirmed by Paz et al. [54]. However, Boni et al. [10] could not prove yessotoxin production by the dinoflagellate, indicating the presence of toxic and nontoxic strains of this species or variability in toxin production.
L. polyedrum regularly forms massive blooms along the coast of Southern California [1, 35, 41, 73], with chlorophyll a concentrations attaining 500 mg/m3 in surface waters and dinoflagellate cell abundances as high as 2 × 107/L [35, 41]. L. polyedrum population dynamics are highly seasonal along this coast, with major blooms in spring and autumn (April/May, September/October, respectively) and maximum abundances of up to 1.3 × 107 cells/L (bloom event in Long Beach, CA, in October 2002, unpublished data).
In spite of its ecological significance, relatively little is known about the role of L. polyedrum in planktonic food webs, i.e., specific factors controlling bloom dynamics or the impact of blooms on trophic relationships in the plankton. A number of studies have investigated different physiological aspects of L. polyedrum with regard to its bioluminescence [16, 30, 52], the effect of turbulence on its growth and division rates [40, 70], its life cycle and resting stages [22, 46, 56], as well as predation on L. polyedrum [37–39, 46]. However, knowledge of the population dynamics of the dinoflagellate and the factors that affect its growth at abundances less than bloom densities are still lacking. This work requires methodology that can accurately count these cells at low densities in natural assemblages.
Monitoring coastal waters for the presence of HAB species to assess the potential for bloom formation presently involves accurate morphological identification and enumeration of phytoplankton species using light or electron microscopy. However, analyses by microscopy are time consuming and require a high level of taxonomic expertise [57]. Alternative approaches that are now being developed involve the use of genetic signatures of various taxa that can be detected using modern molecular techniques [45, 49, 51, 61]. These approaches have been demonstrated to be extremely useful for the accurate identification of specific microorganisms [7, 26, 62, 64]. Among these, quantitative real-time polymerase chain reaction (qPCR) enables species-specific detection and enumeration of particular target species in natural water samples. QPCR techniques estimate the abundance of specific gene sequences in a sample as a convenient proxy for the abundance of a target organism. The method offers extreme sensitivity and specificity and yields accurate estimates of target DNA quantity over several orders of magnitude, thus providing an approach to characterize specific taxa over tremendous ranges of abundance in natural samples.
PCR amplification techniques targeting ribosomal DNA (rDNA) have been successfully employed for the detection of a number of toxic dinoflagellates in seawater samples [11, 15, 24, 55]. Ribosomal RNA genes have been the targets of choice for the development of molecular assays because they are present in high copy number and have both highly conserved and hypervariable “signature” regions [32, 47, 48, 63, 69].
Rollo et al. [60] developed a molecular approach for amplifying a specific fragment of the “GP240 family” DNA of L. polyedrum. However, this approach was not quantitative and has not been pursued by other researchers.
In the present study, a qPCR assay for the detection and enumeration of L. polyedrum was developed, tested, and applied using a Molecular Beacon™ assay (Public Health Research Institute). Primers and a Molecular Beacon™ probe were developed to target unique sequences in the 18S rRNA gene of L. polyedrum. QPCR was validated in the laboratory and applied to cultures and natural water samples from coastal waters off the coast of Los Angeles. The vertical migration of L. polyedrum was studied in a 2-m glass column and in natural water samples at four different sampling sites in coastal waters where red tides have been observed over the past several years. The method allowed the detection and quantification of L. polyedrum over a range of cell abundances spanning six orders of magnitude. The assay provides an extremely useful tool for future investigations of population dynamics of this HAB species.
Methods
Cultures, Cloning, and Sequencing
L. polyedrum was isolated from natural water samples from the coast of Los Angeles, CA. Five to ten cells were picked directly into f/2 medium [25] without silica and grown at 20°C and light conditions of 170 μE m−2 s−1. The full-length eukaryotic small subunit ribosomal RNA gene (18S ssu rDNA) was amplified in two fragments using universal eukaryotic primers (Euk-A and 1055R for the first fragment and 570F and Euk-B for the second fragment; Table 1) to generate products for subsequent cloning and sequencing. Two to five cells of L. polyedrum were concentrated in a volume of 2 μL of f/2 and then used directly in the PCR reaction. Four replicate PCR reactions were run with the following PCR reagents: 0.5 μM of each primer, 1× buffer B (Promega, Madison, WI), 2.5 mM Promega MgCl2, 250 μM Promega dNTPs, 300 ng/μL BSA (Sigma A-7030 [43]), and 2.5 U of Promega Taq in buffer B. Samples were run on a Bio-Rad iCycler (Hercules, CA) using the following thermal protocol: one cycle at 95°C for 2 min, 70 cycles at 95°C for 30 s, 50°C for 30 s, 72°C for 2 min, a final extension at 72°C for 7 min, and a hold at 4°C. After amplification, DNA products were combined, run on an agarose gel, purified, cloned, and sequenced according to Countway et al. [14]. The first fragment of the 18S rDNA was sequenced with the primers M13, 373F, 570F, 570R, and 1055R and the second fragment with the primers 570F, 1055F, 1055R, and Euk-B (Table 1). The full-length 18S rDNA sequence was obtained with nearly complete coverage on both DNA strands (GenBank accession nos. DQ202217–DQ202226).
Probe and Primer Design
Molecular Beacons™ are single-stranded oligonucleotides that form a stem-loop structure [75]. The molecular beacon is labeled with a fluorescent marker at its 5′ end and a quencher at its 3′ end. When the beacon is in the closed hairpin loop configuration, the fluorophore is quenched. Upon binding to the amplicon, the quencher is separated from the fluorophore and the probe fluoresces. The fluorescent signal is measured during the annealing step of each PCR cycle.
All L. polyedrum sequences in the GenBank database (http://www.ncbi.nlm.nih.gov) were downloaded and added to an alignment of the sequences from our cultured L. polyedrum to design a species-specific probe and primer set. Based on those sequences, a Molecular Beacon™ probe and flanking primers were designed using a dedicated software package (“Beacon-Designer 4,” Premier Biosoft International, Palo Alto, CA) that rated both probe and primers as “best” with regard to secondary DNA structure and thermodynamic parameters. A species-specific forward primer (5′-GCTTCGCT TGCTCCTTTGG-3′) was designed to anneal at base 227 of the L. polyedrum 18S rDNA sequence generated from our culture. A nonspecific reverse primer (5′-TCTCCGTTACCCGTCATTGC-3′) annealing at base 366 was selected to create a 140-bp amplification product. A Molecular Beacon™ probe (set apart by brackets in the following sequence) with seven complimentary “stem-forming” nucleotides at the 5′ and 3′ ends was designed to anneal at base 304 of the 18S sequence (5′-CGCGATC[TCTGACCTATCAGCTTCGGATGGTAG]GATCGCG-3′). The specificity of probe and primer sequences was evaluated through comparison to sequences in GenBank, using basic local alignment search tool (BLAST) [2] against the NCBI database [8]. In addition to BLAST comparisons, probe and primer sequences were manually aligned with BioEdit Version 7.0 [27] to sequences of closely related dinoflagellates obtained from the GenBank to examine the degree of mismatch between our primers/probe and closely related dinoflagellates. These public sequences were derived from different species of Scrippsiella, Pheopolykrikos, Peridinium, Prorocentrum, Dinophysis, Gyrodinium, Thoracosphaera, Adenoides, Gymnodinium, Gonyaulax, Takayama, and Pfiesteria.
The forward primer matched three out of the four L. polyedrum sequences from GenBank. The fourth sequence had four mismatches with the forward primer. However, that sequence was only 96–97% similar to the other three sequences identified as L. polyedrum in GenBank (the other three sequences had a similarity of 99% to each other). We did not consider this fourth sequence in subsequent probe and primer design because 3–4% dissimilarity indicated that this fourth sequence was probably not L. polyedrum. The forward primer matched the L. polyedrum sequence of our laboratory culture. The next closest BLAST hits for the forward primer did not hit any other protistan sequences (only vertebrate sequences with one to three mismatches plus two fungi and three bacteria with three mismatches each). The reverse primer was not species-specific but matched dinoflagellates exclusively within the 100 most closely matching BLAST hits. The beacon sequence was specific to all L. polyedrum sequences, but was different from other dinoflagellate and stramenopile sequences at a single nucleotide position. The high-performance liquid chromatography (HPLC)-purified beacon, with a 6-FAM (carboxyfluorescein) reporter on the 5′ end and Black Hole Quencher™-1 (Biosearch Technologies, Inc., Novato, CA) on the 3′ end, and HPLC-purified primers were ordered from Proligo LLC (Boulder, CO).
Probe and Primer Real-Time Testing
Real-time PCR reactions were performed using a Bio-Rad iCycler iQ™ (Bio-Rad) real-time detection system. Different concentrations of Mg2+ were tested and optimized for qPCR using L. polyedrum plasmid DNA containing the target sequence (Fragment: Euk-A-1055R) as a template for amplification. Concentrations of 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, and 6 mM Mg2+ were tested in different PCR reagent master mixes. A Mg2+ concentration of 5.5 mM yielded the highest signal and was used in all subsequent real-time PCR reactions. Total reaction volumes of 50 μL were used for each sample composed of 1 μL of plasmid (3.5 ng/μL) and 49 μL of master mix, consisting of 0.5 μM of each primer, 0.25 μM Beacon, 1× Promega buffer B, 5.5 mM Promega MgCl2, 250 μM Promega dNTPs, 300 ng/μL BSA (Sigma A-7030 [43]), and 2.5 U of Promega Taq in buffer B. Samples were processed on a Bio-Rad iCycler iQ™ using the following thermal protocol: 1 cycle at 95°C for 30 s, 35 cycles at 95°C for 30 s, 55°C for 1 min, 72°C for 30 s, and a hold at 4°C. Specificity of the primers was also tested by examining the fragment generated by PCR using SYBR® Green dye (Molecular Probes, Eugene, OR) to bind to double-stranded DNA during the amplification [33]. A subsequent melt-curve analysis was conducted to ensure the amplification of a product of only one size and to find the best annealing temperature for the primers [12, 71].
Cross-Reactivity Test
The molecular beacon probe and primers were tested in real-time PCR with a cell lysate of L. polyedrum and lysates of 45 nontarget organisms to ascertain the specificity of the assay. Clonal cultures of a variety of other alveolates (dinoflagellates, ciliates), stramenopiles (eight classes), Cryptophytes, Euglenozoa, Haptophytes, and Chlorophytes were grown to high abundances (Table 2). Marine phytoplanktons were grown in K-medium [42] modified with inorganic PO4 at f/2 concentration (36 μM); freshwater phytoplanktons were cultured in DY-IV medium [3]. Heterotrophic protists were grown in sterile seawater or freshwater, amended with sterile yeast extract at a final concentration of 0.005% and two to three sterile rice grains to enrich the natural bacterial biota. After confirming high abundances of all species by light microscopy, a total volume of 250 mL of each culture was collected on a 45-mm GF/F filter (Whatman Inc., Florham Park, NJ). Two milliliters of lysis buffer was added and lysates were created (see below). Lysates were diluted 1:100 in Milli-Q® water (Millipore, Billerica, MA), and 12 μL of each lysate was used in the PCR reaction (see below), which was run for 35 cycles (see thermal protocol and master-mix concentrations above).
Standard Curves
Sensitivity of the qPCR assay was analyzed by performing real-time PCR with serial dilutions of plasmid DNA and cell lysates, ranging over several orders of magnitude. In real-time PCR, reactions are analyzed during the initial exponential phase. The rate of increase of fluorescence in a reaction vessel is directly proportional to the quantity of the target molecule initially present. The threshold cycle parameter (C t) is defined as the fractional cycle number at which the fluorescence crosses a fixed threshold above the baseline (using the default “automatic calculation” setting of the Bio-Rad real-time data collection software). Serial dilutions of the dinoflagellate plasmid containing the target sequence (1:10 at each dilution step) were generated spanning eight orders of magnitude by diluting a plasmid DNA stock (11.6 ng/μL). A volume of 10 μL of each diluent was used in the PCR reactions (+40 μL master mix, see concentrations above). Concentrations in the eight plasmid standards (each in triplicate) ranging from 11.6 to 1.16 × 10−6 ng/μL were used to determine the minimum number of 18S rDNA copies of L. polyedrum necessary for detection in our qPCR assay. DNA concentrations of plasmid standards were converted into 18S rDNA copy number using the molecular weight of the 18S rDNA fragment containing the target sequence (plus vector, pGem T-Easy) and the molecular weights of the different nucleotides (obtained from Integrated DNA Technologies).
It is not possible to relate 18S copy numbers directly to dinoflagellate cell abundances because the copy number of 18S rDNA genes in L. polyedrum is unknown. Thus, use of the plasmid DNA for standardizing the qPCR assay, while convenient, does not easily translate to cell abundance of L. polyedrum. We therefore used cell lysates of known abundances of a L. polyedrum culture to estimate the sensitivity of our qPCR approach with regard to cell numbers (as opposed to gene copy numbers) and to directly correlate signal from the qPCR assay to cell number. The cell concentration of the culture was determined by counting a Lugol's preserved subsample [72] with an inverted microscope (Leica DMIRBE) at 100× magnification. Dilutions spanning six orders of magnitude were generated in triplicate, ranging from 3400 to 0.034 cells/mL. A volume of 250 mL of each cell dilution was collected on 25-mm GF/F filters, resulting in a range of 8.5–8.5 × 105 cells on the filters. Filters were loosely rolled and placed into 15-mL Falcon tubes (Falcon Blue Max™ Jr. Polystyrene Conical Tube). Two milliliters of warm (∼40°C) lysis buffer was added [100 mM Tris (pH 8), 40 mM EDTA (pH 8), 100 mM NaCl, 1% sodium dodecyl sulfate], and the filters were frozen until processed further. Lysates were generated by thawing samples in a water bath at 70°C with 200 μL of 0.5-mm zircon beads (for bead beating) added to each sample. Samples were bead-beaten for 30 s on a vortexer set to highest speed and heated again at 70°C for 5 min, repeating this procedure for a total of three times. Lysates were separated from filter debris by forcing the fluid through a 10-mL syringe, after which the lysates were transferred into 2-mL microcentrifuge tubes and stored at −20°C until processed for qPCR.
Cell lysates were used as templates directly in PCR reactions without further purification to avoid variability and low yields typical of traditional DNA purifications [23]. Lysates were diluted 1:100 with MilliQ water to avoid inhibition of the PCR reaction by components of the lysis buffer (such as EDTA) or interference by cell components. Different volumes of the 1:100 dilution (2.5, 5, 7.5, 10, 12.5, and 15 μL) were tested in PCR reactions to optimize lysate volume (i.e., the volume yielding the highest fluorescent signal, lowest C t value) with no inhibition apparent for the amplification. A volume of 12 μL of the 1:100 dilution matched those criteria and was used in all subsequent PCR reactions.
Triplicate samples of all cell lysate standards (made from a culture with known dinoflagellate abundance) were run in a real-time PCR reaction (50 cycles) with the same concentrations of reagents and thermal protocol described above. Threshold cycles were plotted against the log (number of cells/PCR reaction) to generate standard curves. Standard curves were used to interpret the cell abundances of L. polyedrum in natural samples based on their threshold cycles [cell number = (C T − intercept)/slope].
Variability in the standard curves because of different PCR reactions was examined by creating standard curves from replicate samples of a single lysate. In addition, variability of standard curves associated with different cell lysates was examined by harvesting L. polyedrum at four different times from a batch culture. This procedure was undertaken to examine potential variability of DNA content per cell due to cell division and variability due to cell lysis.
Information from the four standard curves generated from different cell lysates were pooled, and a single regression of all data points was used to generate an “average” standard curve. This average standard curve was used to calculate all cell abundances of L. polyedrum in water samples collected from the laboratory water column and from natural water samples.
PCR Amplification Efficiency Tests
The amplification efficiency of the target sequence in natural seawater samples was tested in real-time PCR by comparing standard curves generated from pure cell cultures of L. polyedrum and cultured dinoflagellates spiked into natural seawater. Known numbers of L. polyedrum cells were spiked into 500 mL of natural (unfiltered) seawater and into 500 mL of 0.2-μm filtered seawater. Cell abundances spanned four orders of magnitude (170–170,000 total cells), and samples were filtered and analyzed in triplicate. Cell lysates were generated and diluted 1:100 with MilliQ water as described above. Samples (12 μL) as well as three negative controls of natural seawater (without the dinoflagellate spike) were run in a real-time PCR reactions (50 cycles; reagent concentrations and thermal protocol as described above). The standard curves generated from the threshold cycles for natural seawater and filtered seawater were compared to detect potential differences in PCR efficiency (because of PCR inhibitors in natural seawater and potential competition for probe and primers from DNA of other nontarget species).
Observation of Vertical Migration of L. polyedrum in the Laboratory
The vertical migratory behavior of L. polyedrum was investigated in the laboratory in a 2-m glass column with a diameter of 11 cm. The column was inoculated with 16 L of f/2 medium [25] and 4 L of a concentrated L. polyedrum culture (1600 cells/mL). An artificial thermocline was established at a depth a 107 cm by cooling the column at its base, whereas the surface of the column was kept at 20°C (optimum culture temperature of L. polyedrum). A light/dark cycle of 11:13 h was applied using a 250-W metal halide plant grow bulb suspended above the column. The L. polyedrum was allowed to acclimate in the column for 1 week, and then samples were collected four to five times each day for a period of 3 days (05:00, 09:00, 12:00, 15:00, and 18:00 at days 1–3; 21:00 at days 2–3) at 20 different depths within the column (2, 4, 6, 8, 10, 12, 20, 30, 40, 50, 60, 70, 80, 90, 96, 100, 104, 108, 112, and 127 cm). Samples (5 mL total) were collected at all depths for counts of L. polyedrum by microscopy, whereas additional 10 mL samples were collected for qPCR at six depths (4, 12, 30, 60, 100, and 112 cm). Samples for microscopy were preserved at a concentration of 10% Lugol's iodine solution [72], and 1–2 mL was counted with an inverted microscope (Leica DMIRBE) at 100× magnification. Samples for qPCR were collected on 25-mm GF/F filters, and cells were lysed in 1 mL of lysis buffer, as described above for the standards. Lysates were diluted 1:100 with MilliQ water, and 12 μL of the dilutions was run in real-time PCR (in triplicate) together with a set of six standards (lysates of a serially diluted culture in triplicate) using protocols described above.
Abundances determined by qPCR were plotted against abundances determined by microscopy. Data were pooled for the sampling times in the morning (05:00–12:00) and in the afternoon and evening (15:00–21:00) because we noted a pattern in the ratio between counts obtained using these methods that related to time of day (see Results). The slopes of resulting regressions were compared with a percentile bootstrap method [80] for significant differences. Furthermore, ratios of abundances determined by microscopy and abundances determined by qPCR were calculated for the pooled data.
Application of qPCR to Natural Water Samples
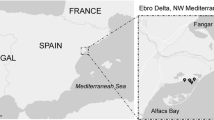
Water samples were collected from shore at Long Beach, CA, and at three different locations off the coast of Los Angeles. Stations were situated along an onshore–offshore transect from Los Angeles Harbor toward Santa Catalina Island into the San Pedro Channel (Fig. 1). This area commonly experiences red tides caused by L. polyedrum. Samples were transferred to the laboratory in a cooler, where they were processed for microscopy and qPCR as described above. Ninety milliliters of the samples was preserved with 10-mL Lugol's iodine solution for microscopy. Samples for qPCR (400 mL) were collected onto 25-mm GF/F filters, loosely rolled into 15-mL Falcon tubes, and 2 mL of lysis buffer was added (see above). Samples were lysed and diluted in the same way as described above and run in triplicate together with six triplicate standards (see above). For microscopy, 10–50 mL of the preserved sample was settled onto slides overnight and counted with an inverted microscope (Leica DMIRBE) at 100× magnification.

Sampling sites off the coast of Los Angeles for detecting and quantifying Lingulodinium polyedrum in natural seawater. LB = Long Beach, S1–S3 = Stations 1–3.
Results
Cross-Reactivity Tests
The cross-reactivity test carried out with lysates of L. polyedrum and 45 nontarget cultures showed significant amplification above the baseline only for L. polyedrum after 35 cycles in the qPCR reaction (Table 2). No signal was obtained for any of the nontarget cultures, supporting the specificity of our Molecular Beacon™ assay for the quantitative detection of L. polyedrum and confirming the results of our previous in silico analysis.
Standard Curves
The standard curves created with plasmid DNA and cell lysates spanned eight and six orders of magnitude, respectively (Fig. 2a,b). The plasmid DNA standard curve ranged from 11.6 to 1.16 × 10−6 ng/μL (Fig. 2a). The plasmid containing the target sequence (including the pGem-T Easy vector) had a molecular weight of 2.7 × 106 Da = 4.4 × 10−18 g. The lowest plasmid concentration that was detected was 1.16 × 10−6 ng/μL. Corrected for the molecular weight of the plasmid (4.4 × 10−18 g), this equates to a detection limit of 264 18S ssu rDNA molecules.

Standard curves created from serial dilutions of plasmid DNA (a) and cell lysates of a serially diluted culture (b, c) of L. polyedrum, plotted as threshold cycles (C t) versus log of the plasmid concentration (ng/μL) or log of cell number per PCR reaction, respectively. The slopes, intercepts, and correlation coefficients (r 2) of linear regressions of the data are shown in (a) and (c). (b) Four standard curves created from the same set of cell lysates analyzed in different qPCR reactions. (c) Standard curve created from the average of four different cell lysates obtained at different time points during the exponential growth phase of a L. polyedrum culture.
The standard curves created with cell lysates of known numbers of L. polyedrum cells ranged from 850,000 to 8.5 cells on the GF/F filter. The regressions of four different qPCR runs using the same lysate were highly similar (Fig. 2b). Greater variability was observed for a regression of four different cell lysates, indicating some variability associated with cell cycle or extraction efficiency (Fig. 2c). The relationship obtained from the regression of these four lysates (Fig. 2c) was used for all subsequent calculations of cell abundance.
The sensitivity of the qPCR assay (8.5 cells total) of course depends on the volume of sample filtered. For a volume of 400 mL (as we typically use for field samples), this corresponds to a sensitivity of approximately 21 cells/L for the method as applied in this study. Many field studies filter as much as 2 L for natural water samples. This volume would increase the sensitivity of the qPCR assay to approximately 4 cells/L assuming no reduction in PCR efficiency.
PCR Amplification Efficiency
The amplification efficiency of the Molecular Beacon™ assay was tested by examining natural unfiltered seawater and seawater filtered through 0.2-μm filters to which a known number of dinoflagellates were added. Filtration should remove any particulate materials present in natural seawater that might inhibit PCR amplification. Standard curves created from lysates of these samples spanned four orders of magnitude and were highly similar (Fig. 3). Natural seawater without added L. polyedrum cells served as a negative control and did not show amplification above threshold values (at 35 cycles), indicating that the abundance of L. polyedrum in the natural seawater was below the limit of detection of the method, and that signals in the standard curves were indicative of the L. polyedrum cells spiked into the seawater. No inhibitory effects were observed from natural seawater on the PCR reaction. This result was evident from nearly identical curves obtained with and without particulate material present. There was also no observable competition for probe or primers with DNA of other species in natural seawater.

Standard curves created from serial dilutions of L. polyedrum cells spiked into natural seawater (SW) and 0.2-μm filtered seawater (FSW). The threshold cycles (C t) were plotted against the log of the cell numbers per PCR reaction. The slopes, intercepts, and correlation coefficients (r 2) of linear regressions of the data are shown.
Vertical Migration of L. polyedrum in the Laboratory
L. polyedrum demonstrated a consistent vertical migratory behavior throughout the 3-day period examined in this study. Cells were highly concentrated in the surface layer of the column (0–10 cm) during the morning. This process began at least 1 h prior to the light coming on (the first morning sample was collected at 05:00, whereas the light went on at 06:00). The vertical distribution of the L. polyedrum population became more uniform throughout the column during the afternoon and was almost evenly dispersed throughout the evening (Fig. 4).

Vertical distributions of cell abundances of L. polyedrum from a water column established in the lab at six sampling times per day, determined by microscopy. Cell abundances were pooled over 3 days of observation and are presented as means and standard errors (SE).
Cell abundances determined via qPCR were compared to abundances determined by microscopy (Figs. 5 and 6). Counts of L. polyedrum by the two methods were in general agreement for some but not all samples. Microscopy-based counts were lower than abundances estimated using the qPCR assay mostly for samples collected in the morning, whereas microscopy-based counts were higher than qPCR estimates in the majority of samples collected during mid and late afternoon (note the deviation of data points from the line of 1:1 correspondence in Fig. 6). Data pooled from samples collected between 5:00 and 12:00 were compared to data from samples collected between 15:00 and 21:00 and were found to differ significantly from each other (p < 0.01). The ratios of these abundances (no. of microscopy/no. of qPCR) were 0.75 ± 0.1 (mean ± standard error) for the morning samples (5:00–12:00) and 1.66 ± 0.23 (mean ± standard error) for the afternoon samples (15:00–21:00).

Cell abundances of L. polyedrum from a water column established in the lab at six sampling times on day 2, determined by microscopy (20 depths sampled) and qPCR (6 depths sampled, mean and standard error of triplicate PCR reactions).

Cell abundances of L. polyedrum from a water column established in the lab determined by microscopy and by qPCR. The solid line indicates a 1:1 relationship between the two methods. The filled symbols represent the morning samples (5:00–12:00), whereas the empty symbols represent the afternoon and evening samples (15:00–21:00).
L. polyedrum in Natural Water Samples
L. polyedrum was detected in October and November 2004 at all four sampling locations in the vicinity of Los Angeles Harbor and Long Beach (Fig. 7). In October, abundances were highest in nearshore waters off Long Beach with maximum abundances approaching 106 cells/L. Abundances observed at the offshore Stations 1–3, when detected, were lower during October compared to the Long Beach samples, but were similar at the different locales during November. L. polyedrum was detected at most sampling times during November at Station 1, whereas the dinoflagellate was detected sporadically during October and November at Stations 2 and 3 (Fig. 7). L. polyedrum was not detected at the Long Beach sampling site from December through the beginning of February (abundances were not determined at Stations 1–3). Overall, microscopy-based counts for the natural samples collected in this study were in close agreement with cell abundances estimated by qPCR (Figs. 7 and 8).

Cell abundances of L. polyedrum determined by qPCR (vertical bars, mean and standard error of triplicate PCR reactions) and microscopy (black diamonds) from field samples near Long Beach and at Stations 1–3 on different sampling dates in 2004/2005. Thin horizontal lines indicate the approximate limit of detection for L. polyedrum via qPCR when analyzing 400-mL samples (21 cells/L). Nd = not detected, nm = not measured, i.e. samples were not analyzed for these time points.

Cell abundances of L. polyedrum determined by microscopy and by qPCR for the different sampling locations (Long Beach, Stations 1–3). The solid line indicates a 1:1 relationship between the two methods.
Discussion
The red tide dinoflagellate L. polyedrum is an important phytoplankter of coastal ecosystems. This organism can have a significant impact on planktonic food webs because of the formation of massive blooms and the potential for toxin production [18, 41, 46, 54]. L. polyedrum is well studied with respect to some aspects of its life cycle and physiology (see review by [46]), but mechanisms and factors leading to bloom formation and demise are still poorly understood.
The timely detection of harmful algal species prior to bloom development represents a critical component of most HAB management plans as well as a requisite tool for researchers studying population dynamics and developing models to forecast HAB events. The qPCR assay described in this study has proven to be specific and sensitive for the detection and quantification of L. polyedrum in both cultures and environmental samples. This approach is based on standard curves created from lysates of known cell numbers and thus allows direct extrapolation of the qPCR results to dinoflagellate abundances. Abundances in unknown samples can be directly calculated from standard curves over a range of six orders of magnitude, ranging from a minimum detection limit of 8.5 to at least 850,000 cells.
The qPCR approach for quantitative detection of L. polyedrum was employed to monitor the dinoflagellate in natural samples at abundances ranging from 106 cells/L in October down to “background” abundances during November. A volume of 400 mL was filtered for these analyses, resulting in a limit of detection of approximately 21 cells/L. This level of sensitivity and the dynamic range of the approach allowed the study of population dynamics of L. polyedrum at the study sites off the coast of Southern California at densities well below bloom abundances. Cell abundances determined in natural samples using our qPCR method agreed well with counts obtained by light microscopy (Fig. 8).
The qPCR method was also employed to follow the population dynamics of L. polyedrum in the laboratory using a 2-m glass column to observe its vertical migratory behavior. L. polyedrum exhibited a strong vertical migration with aggregation in the surface layer of the water column during the morning, less pronounced aggregation in the column during the afternoon, and a relatively even distribution of the population within the column above the thermocline at night. The dinoflagellate began to aggregate near the surface at least 1 h before the lights came on. Circadian rhythms for many major cell functions of L. polyedrum have been demonstrated including bioluminescence, photosynthetic activity, swimming behavior, and cell division (see reviews by [50, 59]). Diel vertical migration has also been observed in L. polyedrum in the laboratory [20, 31] and in the field [9, 21, 77]. Our observation that upward migration begins before the initiation of the light phase implies a circadian rhythm in this dinoflagellate as has been reported for other dinoflagellates in laboratory studies (e.g., [20, 53, 79]). This common pattern among dinoflagellates has been interpreted as a compromise between the need to obtain growth-limiting nutrients (descent at night) and sufficient light for photosynthesis (ascent during the morning) [9, 76]. Our experiment was conducted with nutrient replete conditions throughout the water column (f/2 medium). Therefore, migratory behavior presumably was not influenced by differences in nutrient concentration between surface and deeper waters.
The water column that we established in the laboratory provided a useful system for comparing counts of L. polyedrum based on microscopical, morphology-based identification and by qPCR. Overall, microscopy-based counts were in 1:1 correspondence with estimates obtained using the qPCR assay (Fig. 6). However, microscopy-based counts were lower than abundances determined by qPCR for the morning samples, whereas they were higher for the afternoon samples. We speculate that these differences were a consequence of synchronous growth of L. polyedrum cells in the water column implied by the observed pattern in the ratio between the two counting methods related to the time of day. Samples collected from the column during the afternoon and evening (15:00–21:00) had ratios of microscopical counts to qPCR counts that averaged 1.66, relative to samples collected during the morning (05:00–12:00; average ratio = 0.75). These data imply that the cells in the column contained more DNA per cell during the morning and less DNA per cell during the evening relative to cells harvested for the standard curves. A comparison of the linear regression of these two subsets of the water column data yielded significant differences. These differences presumably reflected the per-cell DNA content of the dinoflagellate in the column in different phases of its cell cycle over the course of the day.
Cell division in L. polyedrum is controlled by a circadian clock [13, 36], which, in turn, is influenced by surrounding light conditions. In the 2-m glass column, the light intensity decreased dramatically from the top to the bottom of the column. Cells migrated vertically presumably to optimize light availability during the photoperiod. This dominant behavior may have led to synchronous cell division. In contrast, our laboratory cultures were maintained in 1-L volumes under bright even illumination, and no evidence of synchronous cell division was observed (data not shown). Interestingly, microscopical counts and qPCR estimates for natural samples analyzed in this study were in close agreement (Fig. 8), presumably indicating the absence of synchronous cell division in the natural water samples.
The qPCR method that we report here has several major advantages relative to traditional morphology-based (microscopy) methods. These include high sensitivity of the method, broad dynamic range over which cell abundance can be quantified (at least six orders of magnitude), rapidity of the analysis, and freedom from the requirement of morphological taxonomic expertise for the accurate identification of L. polyedrum. The qPCR approach dramatically improves sensitivity and dynamic range in analyzing natural samples for L. polyedrum.
Our specific approach as applied to field samples entailed the analysis of 400-mL samples, resulting in a limit of detection of approximately 21 cells/L. This detection limit might be reduced by filtering a larger volume of water, although, at some point, PCR poisons will cause a reduction in signal. Many molecular analyses employ filtration of 2 L of water, which would increase the sensitivity of the qPCR approach to 4 cells/L. This ability to accurately identify low abundances of L. polyedrum is complemented with a very large dynamic range. Samples of all concentrations (over six orders of magnitude) can be analyzed on a single plate using the same amount of cell lysate. Sample volumes for microscopy must be adjusted for the abundance of the target species. Concentrated samples need to be diluted, whereas samples with low L. polyedrum abundances need to be counted in higher volumes or even in multiple subsamples. These complications mean that the analysis of 20 samples may take as much as a week to fully process. In contrast, our qPCR method provides for the analysis of a set of 18 standards (six standards in triplicate) and 78 unknown samples (26 unknowns in triplicate) on a single 96-well plate within approximately 5 h (including preparation of cell lysates of unknowns and a qPCR run with 35 cycles).
We have developed and validated a qPCR approach for the detection and quantification of L. polyedrum in natural samples. The high sensitivity and broad dynamic range of the method provides a useful new tool for studying bloom initiation by this red-tide-forming dinoflagellate. This new approach will allow studies of population dynamics within mixed assemblages facilitating new insights into the factors governing the growth and trophic dynamics of this dinoflagellate.
References
Allen, WE (1946) “Red water” in La Jolla Bay in 1945. Trans Am Microsc Soc 65: 262–264
Altschul, SF, Madden, TL, Schaffer, AA, Zhang, J, Zhang, Z, Miller, W, Lipman, DJ (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402
Andersen, RA, Morton, SL, Sexton, JP (1997) Provasoli-Guillard National Center for Culture of Marine Phytoplankton 1997 list of strains. J Phycol 33(Suppl): 1–75
Anderson, DM (1995) Toxic red tides and harmful algal blooms: A practical challenge in coastal oceanography. Rev Geophys 33: 1189–1200
Anderson, DM, Gilbert, PM, Burkholder, JM (2002) Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries 25: 704–726
Anderson, DM, Keafer, BA, Geyer, WR, Signell, RP, Loder, TC (2005) Toxic Alexandrium blooms in the western Gulf of Maine: The plume advection hypothesis revisited. Limnol Oceanogr 50:328–345
Anderson, DM, Kulis, DM, Keafer, BA, Berdalet, E (1999) Detection of the toxic dinoflagellate Alexandrium fundyense (Dinophyceae) with oligonucleotide and antibody probes: Variability in labeling intensity with physiological condition. J Phycol 35: 870–883
Benson, DA, Karsch-Mizrachi, I, Lipman, DJ, Ostell, J, Wheeler, DL (2004) GenBank: Update. Nucleic Acids Res 32: D23–D26
Blasco, D (1978) Observations on the diel migration of marine dinoflagellates off the Baja California Coast. Mar Biol (N Y) 46: 41–47
Boni, L, Ceredi, A, Guerrini, F, Milandri, A, Pistocchi, R, Poletti, R, Pompei, M (2001) Toxic Protoceratium reticulatum (Peridiniales, Dinophyta) in the north-western Adriatic sea (Italy). In: Hallegraef, GM, Blackburn, SI, Bold, CI, Lewis, RI (Eds.) Harmful Algal Blooms 2000, Intergovernmental Oceanographic Commission of UNESCO, pp 137–140
Bowers, HA, Tengs, T, Glasgow Jr, HB, Burkholder, JM, Rublee, PA, Oldach, PA (2000) Development of real-time PCR assays for rapid detection of Pfiesteria piscicida and related dinoflagellates. Appl Environ Microbiol 66: 4641–4648
Bustin, SA, Nolan, T (2004) Chemistries. In: Bustin, SA (Ed.) A–Z of quantitative PCR, 5. International University Line. La Jolla, CA, pp 215–278
Cetta, CM, Anderson, DM (1990) Cell cycle studies of the dinoflagellates Gonyaulax polyedra Stein and Gyrodinium uncatenum Hulburt during asexual and sexual reproduction. J Exp Mar Biol Ecol 135: 69–83
Countway, PD, Gast, RJ, Savai, P, Caron, DA (2005) Protistan diversity estimates based on 18S rDNA from seawater incubations in the Western North Atlantic. J Eukaryot Microbiol 52: 95–106
Coyne, KJ, Hutchins, DA, Hare, CE, Cary, SC (2001) Assessing temporal and spatial variability in Pfiesteria piscicida distributions using molecular probing techniques. Aquat Microb Ecol 24: 275–285
Dassow, Pv, Bearon, RN, Latz, MI (2005) Bioluminescent response of the dinoflagellate Lingulodinium polyedrum to developing flow: Tuning of sensitivity and the role of desensitization in controlling a defensive behavior of a planktonic cell. Limnol Oceanogr 50: 607–619
Dodge, JD (1989) Some revisions of the family Gonyaulacaceae (Dinophyceae) based on a scanning electron microscope study. Bot Mar 32: 275–298
Draisci, R, Ferrettii, E, Palleschi, L, Marchiafava, C, Poletti, R, Vivani, R (1999) High levels of yessotoxin in mussels and presence of yessotoxin and homo-yessotoxin in dinoflagellates of the Adriatic Sea. Toxicon 37: 1187–1193
Elwood, HJ, Olsen, GJ, Sogin, ML (1985) The small-subunit ribosomal RNA gene sequences from hypotrichous ciliates Oxytricha nova and Stylonychia pustulata. Mol Biol Evol 2: 399–410
Eppley, RW, Holm-Hansen, O, Strickland, JDH (1968) Some observations on the vertical migration of dinoflagellates. J Phycol 4: 333–340
Eppley, RW, Reid, FMH, Cullen, JJ, Winant, CD, Stewart, E (1984) Subsurface patch of a dinoflagellate (Ceratium tripos) off Southern California: Patch length, growth rate, associated vertically migrating species. Mar Biol (N Y) 80: 207–214
Figueroa, RI, Bravo, I (2005) Sexual reproduction and two different encystment strategies of Lingulodinium polyedrum (Dinophyceae) in culture. J Phycol 41: 370–379
Galluzzi, L, Penna, A, Bertozzini, E, Vila, M, Garces, E, Magnani, M (2004) Development of a real-time PCR assay for rapid detection and quantification of Alexandrium minutum (a Dinoflagellate). Appl Environ Microbiol 70: 1199–1206
Godhe, A, Otta, SK, Rehnstam-Holm, A-S, Karunasagar, I, Karunasagar, I (2001) Polymerase chain reaction in detection of Gymnodinium mikimotoi and Alexandrium minutum in field samples from Southwest India. Mar Biotechnol 3: 152–162
Guillard, RRH, Ryther, JH (1962) Studies of marine planktonic diatoms. I. Cyclotella nana Hustedt and Detonula confervacea. Cleve Can J Microbiol 8: 229–239
Guillou, L, Nézan, E, Cueff, V, Erard-Le Denn, E, Cambon-Bonavita, M-A, Gentien, P, Barbier, G (2002) Genetic diversity and molecular detection of three toxic dinoflagellate genera (Alexandrium, Dinophysis, and Karenia) from French coasts. Protist 153: 223–238
Hall, TA (1999) BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98
Hallegraeff, GM (1993) Review of harmful algal blooms and their apparent global increase. Phycologia 32: 79–99
Hallegraeff, GM, Bolch, CJ (1992) Transport of diatom and dinoflagellate resting spores in ships’ ballast water: Implications for plankton biogeography and aquaculture. J Plankton Res 14: 1067–1084
Hastings, JW, Sweeney, BM (1960) The action spectrum for shifting the phase of the rhythm of luminescence in Gonyaulax polyedra. J Gen Physiol 43: 697–705
Heaney, SI, Eppley, RW (1981) Light, temperature and nitrogen as interacting factors affecting diel vertical migrations of dinoflagellates in culture. J Plankton Res 3: 331–344
Hershkovitz, MA, Lewis, LA (1996) Deep-level diagnostic value of the rDNA-ITS region. Mol Biol Evol 13: 1276–1295
Higuchi, R, Dollinger, G, Walsh, PS, Griffith, R (1992) Simultaneous amplification and detection of specific DNA sequences. Bio/Technology 10: 413–417
Hoagland, P, Anderson, DM, Kaoru, Y, White, AW (2002) The economic effects of harmful algal blooms in the United States: Estimates, assessment issues, and information needs. Estuaries 25: 819–837
Holmes, RW, Williams, PM, Eppley, RW (1967) Red water in La Jolla Bay, 1964–1966. Limnol Oceanogr 12: 503–512
Homma, K, Hastings, JW (1988) Cell cycle synchronization of Gonyaulax polyedra by filtration: Quantized generation times. J Biol Rhythms 3: 49–58
Jeong, HJ, Kim, SK, Kim, JS, Kim, ST, Yoo, YD, Yoon, JY (2001) Growth and grazing rates of the heterotrophic dinoflagellate Polykrikos kofoidii on red-tide and toxic dinoflagellates. J Eukaryot Microbiol 48: 298–308
Jeong, HJ, Shim, JH, Kim, JS, Park, JY, Lee, CW, Lee, Y (1999) Feeding by the mixotrophic thecate dinoflagellate Fragilidium cf. mexicanum on red-tide and toxic dinoflagellates. Mar Ecol Prog Ser 176: 263–277
Jeong, HJ, Yoon, JY, Kim, JS, Yoo, YD, Seong, KA (2002) Growth and grazing rates of the prostomatid ciliate Tiarina fusus on red-tide and toxic algae. Aquat Microb Ecol 28: 289–297
Juhl, AR, Velazquez, V, Latz, MI (2000) Effect of growth conditions on flow-induced inhibition of population growth of a red-tide dinoflagellate. Limnol Oceanogr 45: 905–915
Kahru, M, Mitchell, BG (1998) Spectral reflectance and absorption of a massive red tide off Southern California. J Geophys Res Oceans 103: 21601–21609
Keller, DK, Selvin, RC, Claus, W, Guillard, RRL (1987) Media for the culture of oceanic ultraphytoplankton. J Phycol 23: 633–638
Kirchman, DL, Yu, L, Fuchs, BM, Amann, R (2001) Structure of bacterial communities in aquatic systems as revealed by filter PCR. Aquat Microb Ecol 26: 13–22
Kofoid, CA (1911) Dinoflagellata of the San Diego region. IV. The genus Gonyaulax, with notes on its skeletal morphology and a discussion of its generic and specific characters. Univ Calif Publ Zool 8: 187–286
LaJeunesse, TC (2001) Investigating the biodiversity, ecology, and phylogeny of endosymbiotic dinoflagellates in the genus Symbiodinium using the ITS region: In search of a “species” level marker. J Phycol 37: 866–880
Lewis, J, Hallett, R (1997) Lingulodinium polyedrum (Gonyaulax polyedra) a blooming dinoflagellate. Oceanogr Mar Biol: Annu Rev 35: 97–161
Medina, M, Collins, AG, Silberman, JD, Sogin, ML (2001) Evaluating hypotheses of basal animal phylogeny using complete sequences of large and small subunit rRNA. Proc Natl Acad Sci U S A 98: 9707–9712
Medlin, L, Elwood, HJ, Stickel, S, Sogin, ML (1988) The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71: 491–499
Medlin, L, Lange, M, Wellbrock, U, Donner, G, Elbrächter, M, Hummert, C, Luckas, B (1998) Sequence comparisons link toxic European isolates of Alexandrium tamarense from the Orkney Islands to toxic North American stocks. Eur J Protistol 34: 329–335
Mittag, M, Hastings, JW (1996) Exploring the signaling pathway of circadian bioluminescence. Physiol Plant 96: 727–732
Moon-van der Staay, SY, De Wachter, R, Vaulot, D (2001) Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature 409: 607–610
Nicolas, M, Nicolas, G, Johnson, C, Bassot, J, Hastings, J (1987) Characterization of the bioluminescent organelles in Gonyaulax polyedra (dinoflagellates) after fast-freeze fixation and antiluciferase immunogold staining. J Cell Biol 105: 723–735
Park, JG, Jeong, MK, Lee, JA, Cho, KJ, Kwon, OS (2001) Diurnal vertical migration of a harmful dinoflagellate, Cochlodinium polykrikoides (Dinophyceae), during a red tide in coastal waters of Namhae Island, Korea. Phycologia 40: 292–297
Paz, B, Riobo, P, Fernandez, AL, Fraga, S, Franco, JM (2004) Production and release of yessotoxins by the dinoflagellates Protoceratium reticulatum and Lingulodinium polyedrum in culture. Toxicon 44: 251–258
Penna, A, Magnani, M (1999) Identification of Alexandrium (Dinophyceae) species using PCR and rDNA-targeted probes. J Phycol 35: 615–621
Persson, A (2000) Possible predation of cysts—A gap in the knowledge of dinoflagellate ecology. J Plankton Res 22: 803–809
Rehnstam-Holm, AS, Godhe, A, Andersen, AM (2002) Molecular studies of Dinophysis (Dinophyceae) species from Sweden and North America. Phycologia 41: 348–357
Reid, PC (1997) Discharges from hydroelectric power schemes as a trigger for marine algal blooms. Mar Pollut Bull 34: 730–733
Roenneberg, T (1996) The complex circadian system of Gonyaulax polyedra. Physiol Plant 96: 733–737
Rollo, F, Sassaroli, S, Boni, L, Marota, I (1995) Molecular typing of the red-tide dinoflagellate Gonyaulax polyedra in phytoplankton suspensions. Aquat Microb Ecol 9: 55–61
Rubio, MB, Hermosa, MR, Keck, E, Monte, E (2005) Specific PCR assays for the detection and quantification of DNA from the biocontrol strain Trichoderma harzianum 2413 in soil. Microb Ecol 49: 25 – 33
Saito, K, Drgon, T, Robledo, JAF, Krupatkina, DN, Vasta, GR (2002) Characterization of the rRNA locus of Pfiesteria piscicida and development of standard and quantitative PCR-based detection assays targeted to the nontranscribed spacer. Appl Environ Microbiol 68: 5394–5407
Scholin, CA, Hallegraeff, GM, Anderson, DM (1995) Molecular evolution of the Alexandrium tamarense “species complex” (Dinophyceae): Dispersal in the North American and West Pacific regions. Phycologia 34: 472–485
Scholin, CA, Marin, R, Miller, PE, Doucette, GJ, Powell, CL, Haydock, P, Howard, J, Ray, J (1999) DNA probes and a receptor-binding assay for detection of Pseudo-Nitzschia (Bacillariophyceae) species and domoic acid activity in cultured and natural samples. J Phycol 35: 1356–1367
Smayda, TJ (1990) Novel and nuisance phytoplankton blooms in the sea: Evidence for a global epidemic. In: Granéli, E, Sundström, L, Edler, L, Anderson, DM (Eds.) Toxic Marine Phytoplankton. Elsevier. New York, pp 29–40
Smayda, TJ (1997) Harmful algal blooms: Their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnol Oceanogr 42: 1137–1153
Smayda, TJ, Reynolds, CS (2001) Community assembly in marine phytoplankton: Application of recent models to harmful dinoflagellate blooms. J Plankton Res 23: 447–461
Steidinger, KA, Tangen, A (1996) Dinoflagellates. In: Tomas, CR (Ed.) Identifying Marine Diatoms and Dinoflagellates. Academic Press. San Diego, CA, pp 387–584
Stryer, L (1995) Genes for ribosomal RNAs are tandemly repeated several hundred times. In: Stryer, L (Ed.) Biochemistry. W.H. Freeman and Company. New York, NY, pp 992–993
Sullivan, JM, Swift, E, Donaghay, PL, Rines, JEB (2003) Small-scale turbulence affects the division rate and morphology of two red-tide dinoflagellates. Harmful Algae 2: 183–199
Theaker, J (2004) Genotyping using MGB-hydrolysis probes. In: Bustin, SA (Ed.) A–Z of Quantitative PCR, 5. International University Line. La Jolla, CA, pp 733–765
Throndsen, J (1978) Preservation and storage. In: Sournia, A (Ed.) Phytoplankton Manual. UNESCO. Paris, pp 69–74
Torrey, HB (1902) An unusual occurrence of dinoflagellata on the California coast. Am Nat 36: 187–192
Tubaro, A, Sidari, L, Della Loggia, R, Yasumoto, T (1998) Occurrence of yessotoxin-like toxins in phytoplankton and mussels from northern Adriatic Sea. In: Reguera, B, Blanco, J, Fernández, ML, Wyatt, T (Eds.) Harmful Algae. Xunta de Galicia and IOC-UNESCO. Santiago de Compostela, Spain, pp 470–472
Tyagi, S, Kramer, FR (1996) Molecular Beacons: probes that fluoresce upon hybridization. Nat Biotechnol 14: 303–308
Villarino, ML, Figueiras, FG, Jones, KJ, Alvarezsalgado, XA, Richard, J, Edwards, A (1995) Evidence of in-situ diel vertical migration of a red-tide microplankton species in Ria de Vigo (Nw Spain). Mar Biol (N Y) 123: 607–617
Walsh, JJ, Kelley, JC, Whitledge, TE, MacIsaac, JJ, Huntsman, SA (1974) Spin-up of the Baja California upwelling ecosystem. Limnol Oceanogr 19: 553–572
Weekers, PHH, Gast, RJ, Fuerst, PA, Byers, TJ (1994) Sequence variations in small-subunit ribosomal RNAs of Hartmannella vermiformis and their phylogenetic implications. Mol Biol Evol 11: 684–690
Weiler, CS, Karl, DM (1979) Diel changes in phased-diving cultures of Ceratium furca (Dinophyceae): Nucleotide triphosphates, adenylate energy charge, cell carbon, and patterns of vertical migration. J Phycol 15: 384–391
Wilcox, RR (2003) Applying Contemporary Statistical Techniques Elsevier Science (USA). Academic Press. San Diego, USA
Yanisch-Perron, C, Vieira, J, Messing, J (1985) Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 33: 103–119
Acknowledgments
We thank R. A. Schaffner, J. M. Rose, and A. Schnetzer (USC, Los Angeles) for helpful discussions and help with graphics and statistics, the Centre for Embedded Network Sensing (CENS) for providing the glass column for our laboratory work, and the Deutscher Akademischer Austauschdienst (DAAD) for their financial support. This project was additionally supported by National Science Foundation grants MCB-0084231, OPP-0125437, The Centre for Embedded Networked Sensing (CENS) under the NSF Cooperative Agreement CCR-0120778, and Environmental Protection Agency grant RD-83170501.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moorthi, S.D., Countway, P.D., Stauffer, B.A. et al. Use of Quantitative Real-Time PCR to Investigate the Dynamics of the Red Tide Dinoflagellate Lingulodinium polyedrum . Microb Ecol 52, 136–150 (2006). https://doi.org/10.1007/s00248-006-9030-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-006-9030-3