
Abstract
Forestry practises such has drainage have been shown to decrease emissions of the greenhouse gas methane (CH4) from peatlands. The aim of the study was to examine the methanogen populations in a drained bog in northern Finland, and to assess the possible effect of ash fertilization on potential methane production and methanogen communities. Peat samples were collected from control and ash fertilized (15,000 kg/ha) plots 5 years after ash application, and potential CH4 production was measured. The methanogen community structure was studied by DNA isolation, PCR amplification of the methyl coenzyme-M reductase (mcr) gene, denaturing gradient gel electrophoresis (DGGE), and restriction fragment length polymorphism (RFLP) analysis. The drained peatland showed low potential methane production and methanogen diversity in both control and ash-fertilized plots. Samples from both upper and deeper layers of peat were dominated by three groups of sequences related to Rice cluster-I hydrogenotroph methanogens. Even though pH was marginally greater in the ash-treated site, the occurrence of those sequences was not affected by ash fertilization. Interestingly, a less common group of sequences, related to the Fen cluster, were found only in the fertilized plots. The study confirmed the depth related change of methanogen populations in peatland.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The increasing concentration of atmospheric methane contributes to global warming. Wetlands, including peatlands, are the main source of natural methane emission [18] because anoxic layers of peat provide habitat for methanogenic Archaea (kingdom Euryarchaeota). They produce methane during the final step of the degradation of organic matter.
About one-third of the Finnish land area used to be covered by peatlands, and the proportion of land covered by peatlands is still greater than in any other country [1, 27]. Extensive draining of peatlands in Finland occurred in the 1960s and 1970s to promote tree growth. In addition, ∼1.7 million ha of drained peatlands have been fertilized with ash during the past 50 years [2]. Ash adds nutrients, except nitrogen and phosphorus, and transforms vegetation composition and promotes tree growth [33]. It affects soil microbiota [8, 13] and ectomycorrhiza [13, 31]. Drainage has been shown to decrease methane emissions, the main reasons for the decrease being changes of quantity and quality of substrates for methanogenesis and increased methane oxidation [21, 32]. Methanogen communities in peatlands have been described in undisturbed bogs [14, 17, 36] and fens [9, 10], but the effect of forest management such as ash fertilization on the methane-producing Archaea has, however, never been studied.
We previously studied methanogen communities in an undisturbed fen using specific primers for methanogen 16S rDNA [9] and for the A subunit of the methyl coenzyme-M reductase (mcrA) gene [10]. Mcr, a key enzyme in methanogen metabolism, catalyzes the reduction of methyl coenzyme-M with methane being the final released product [6] and appears to be unique to methanogens [44]. Part of the amino acid sequence is highly conserved [14, 46], and mcr sequence data have consequently been used for estimating methanogen diversity in a variety of environments [5, 28–30, 36, 45].
In this study, we examined possible effects of ash fertilisation on methanogen community structure in ash-fertilized (15,000 kg/ha) and control (no ash addition) plots of a drained bog. Potential CH4 production was measured in vitro from replicate peat samples taken at different depths. Methanogen population diversity was analysed by using molecular methods (PCR-DGGE, cloning and sequencing) targeting the A subunit of the methyl coenzyme-M reductase gene.
Methods
Experimental Site and Sample Collection
The experimental field was located in Muhos, Pelso, Resula (64°30′N, 26°18′E). The site is within the boreal coniferous zone classified as a Cotton grass pine bog [24]. Ditching was done in the 1930’s and continued in 1994 and 1997 [33]. The bog is in a transforming state where the typical bog vegetation is changing to forest vegetation as a result of drainage [38]. Peat depth averages 100 cm. Pines (Pinus sylvestris) 1–7 m height and 20–60 years of age are growing in the bog. They are intermixed with slowly growing birches (Betula pendula).
Three replicate plots (30 m × 30 m) were treated with 15,000 kg/ha of fly ash (F) in June 1997. Another three replicate plots of the same size were kept unfertilized as control (C). One peat core was taken from each replicate plot with a box sampler (4 × 6.8 × 100 cm). In May 2002 after snow melting, peat samples for potential methane production measurements and for DNA extraction were taken from 10 cm (±2 cm) depth intervals in the anoxic parts of the peat cores (Table 1).
Measurement of Potential Methane Production and pH, and DNA Extraction
Fifteen mL of peat was added to 100-mL, oxygen-free, infusion bottles containing 30 mL of distilled water. The bottles were flushed with 99.96% nitrogen in order to obtain anoxic conditions [20] and sealed with butyl rubber septa. Incubation and gas chromatographic analysis of CH4 were performed as described [10].
pH was measured once a day for 3 days from 10 mL of peat mixed with 20 mL of H2O, and incubated at room temperature (∼23°C).
DNA was extracted from 0.25 g of peat by chemical (detergent) and mechanical (bead beating) cell lysis with the Soil DNA Isolation Kit (Mo Bio Laboratories, Inc., Solana Beach, CA, USA), following the manufacturer’s suggested protocol. The DNA quality and yield was assessed by running the extracted DNA in agarose gel. This extraction method has been shown to provide high reproducibility and efficiency when compared with other methods [37].
PCR Amplification
The mcrA gene was used as molecular marker to study the methanogen diversity. The primer pair ME1-ME2 [14] was used to amplify specifically a 760-bp-long region of the mcrA. A 40-nucleotide-long GC-clamp was added to the forward primer to enable DGGE analyses [35]. The 50-μL PCR mixture contained 25 pmol of the appropriate primer pairs, 200 μM dNTPs, 1 U Red Hot polymerase (Advanced Biotechnologies, Epsom, UK), PCR reaction buffer and 3 μL of template (concentration determined empirically). Bovine serum albumin (0.1 μg/μL) was used to prevent PCR inhibition. The reaction conditions were 30 cycles of 94°C for 45 s, 50°C for 45 s, and 72°C for 2 min. The PCR was performed using a Gene Amp thermal cycler (PerkinElmer, Wellesley, MA, USA). Products were analyzed on 1% agarose gels with ethidium bromide staining.
Cloning, RFLP, Sequencing of PCR Product, and DGGE Fingerprinting
All together eight gene libraries were constructed using DNA extracted and amplified from two depths of the control and fertilized plots. Two replicate libraries were obtained from high (H) potential methane producing layers (10 or 20 cm below water level), and two replicate libraries were obtained from low (L) potential methane producing layers (30 or 40 cm below water level).
PCR products obtained with ME primers without GC-clamp were analyzed by gel electrophoresis, purified with Wizard SV Gel and PCR Clean-Up System (Promega, Mannheim, Germany), and cloned in pGEM-T vector plasmid using JM109 competent cells (Promega) according to the manufacturer’s instructions. Colonies dispersed in H2O were used as templates to amplify inserted mcr sequences with ME primers. PCR products were digested with MspI for RFLP analyses [10].
Colonies showing new distinct fingerprint patterns were selected for sequencing. Plasmid DNA was isolated with the Wizard Mini-Preps (Promega) and sequenced using the vectors universal primers.
The mcr sequences obtained in this study were deposited in the EMBL database under accession nos. AJ586241–AJ586250.
DGGE was performed as described earlier [10] on samples from the high and low potential methane-producing layers of the bog with a D-code system (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions. The denaturing gradient was determined by empirical trials; it ranged from 45 to 70%. The gel was run at 60°C, at a constant voltage of 250 V for 6 h. The bands position was marked manually with gel documentation software Gel Doc 2000 (Bio-Rad). In order to relate the results obtained by DGGE fingerprinting with those from RFLP analysis of clone libraries, bands from DGGE fingerprinting were cut out from the gel, reamplified, and subjected to RFLP analysis. Additionally, cloned mcr sequences representing each RFLP groups were reamplified with GC-clamp and run in DGGE.
Statistical Analysis
Two-way ANOVAs were used to test differences in pH, potential CH4 production, and diversity indices as a function of fertilization treatment and depth. P values were considered significant at the P < 0.1 level.
RFLP data were converted to a distance matrix using the Chord algorithm for abundance data and represented in a dendrogram. DGGE banding patterns were converted to a binary matrix where the presence or absence of a band in each lane at a same height was reported in the matrix with 1 and 0, respectively [9]. The binary matrix was transformed to a distance matrix using Jaccard’s coefficient for presence–absence data, and the resulting similarity values were analyzed by cluster analysis. The percentage of coverage of the clone libraries was estimated, following Good’s methods [11], as (1 − (n/N)) × 100, where n is the number of unique clones detected in a sample of size N.
The Shannon–Weaver diversity index [40] was calculated as:
p i is the proportion of clones belonging to the ith OTU and s is the total number of OTUs; RFLP patterns were considered as operational taxonomic units (OTUs) [34]. Additionally, Fisher’s Alpha log series richness index [7] was calculated. Cluster analysis, rarefaction, and diversity analysis were performed with the PAST package (v.0.97, [15]).
Phylogenetic Analysis
Sequences were compared to those in the EMBL database using the BLAST server [3] at the European Bioinformatics Institute (EBI) (URL http://www.ebi.ac.uk; Hinxton Hall, Cambridge, UK). Amino acid sequences were inferred from the mcr nucleotide sequences. Part of the restriction groups detected in the Pelso-Resula drained bog had been detected earlier in a natural fen [10]; they were designated with capital letters whereas new sequences were designated with numbers. Sequences of approximately 250 amino acids were aligned using the CLUSTAL W package [16] and checked manually. Chimeras were excluded from analysis. A phylogenetic tree was estimated using the PHYLIP package (v.3.57c; J. Felsenstein, University of Washington, http://evolution.genetics.washington.edu/phylip.html). Bootstrap values for 100 replicates were generated with SEQBOOT. PROTDIST was used to compute a distance matrix with Dayhoff PAM 001 as amino acid replacement model. The distance matrix was estimated by FITCH with global rearrangement of branches and randomized species input order. The dendrogram was verified by neighbor-joining and maximum parsimony methods.
Results
pH and Water Table
Peat pH varied between 3.8 and 4.0 among depths in control plots and between 3.9 and 4.2 in ash fertilized plots (Table 1) and was marginally greater in the ash treated site than in the controls (P = 0.105). No differences in pH were observed between depths. The water table was 9 cm deeper in the fertilized plots than in the controls (Table 1).
Potential CH4 Production
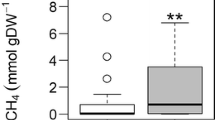
The peat samples taken at the beginning of the growing season in May showed low potential rates of methane production, but CH4 production was detected at all depths (Table 1). At both sites, rates were greater 10 and 20 cm below the water table (upper layer) than 30 and 40 cm below water table (deeper layer). No significant difference in CH4 production was observed between control and fertilized plots.
RFLP of Cloned mcr Sequences
Restriction fragments resulting from the digestion of the mcr gene with MspI were grouped depending on their restriction pattern, and each group was considered as an operational taxonomic unit (OTU). It has previously been shown that sequences grouping in the same OTU have high similarity [10]. Eighteen different OTUs were observed. The percentages of coverage of the clone libraries ranged from 93% to 98% (Table 2), and the richness curves flattened toward an asymptote (Fig. 1). Therefore the number of clones analyzed covered RFLP pattern richness of the libraries. Upper-layer peat from both fertilized and control clone libraries was dominated by sequences L and B, representing between 65% and 95% of all clones. At both sites, libraries from the deeper layers were dominated by one single sequence (E), which constituted 60 to 80% of the deep-layer clones. Some mcr sequences (T, II, III) were practically only found in the fertilized plots (Fig. 2). Cluster analyses of RFLP patterns revealed no ash treatment effect. The libraries did not group by fertilization treatment (Fig. 3a). A depth-related variation of the sequences could clearly be seen. Restriction groups characterizing the upper layers (L, B) were different from the ones characterizing the deeper layers (E) (Fig. 3), and the cluster analysis showed grouping by depth (Fig. 3a). Sample Fb(H) differed from other upper layer samples; it was dominated by pattern E and grouped consequently with deeper layer samples.

RFLP pattern richness curve of eight mcr clone libraries. Curves were obtained by rarefaction calculation. See Methods for designations.

RFLP analysis of mcr clone libraries. Sequences were obtained from control plots (C) and fertilized plots (F), from high (H) and low (L) potential CH4-producing layers of the drained bog. Clone libraries were constructed from replicate peat profiles (a, b, or c). Letters in the legend represent the different RFLP groups. Their phylogenetic affiliation to fen cluster (FC) or rice cluster-I (RC) is indicated in parentheses.

Dendrogram representing the relation between different mcr sequences analyzed by RFLP (a) and DGGE (b). Distances were calculated with Chord’s algorithm for the clone libraries and with Jaccard’s coefficient for DGGE fingerprinting. Samples are from control plots (C) and fertilized plots (F), from high (H) and low (L) potential CH4-producing layers of the drained bog. Different replicate peat profiles (a, b, and c) are represented.
DGGE Fingerprinting
DGGE analysis was performed on replicate samples from control and fertilized sites. A low number of mcr DNA fragments was present in the gels. Cluster analysis of the banding patterns showed no difference in methanogen mcr sequences between control and fertilized plots (Fig. 3b). However, the DGGE fingerprinting revealed a depth-related occurrence of mcr sequences; cluster analysis showed that samples grouped according to depth (Fig. 3b).
RFLP analyses of DGGE bands excised from the gel showed that sequences dominating the clone libraries (B, L, and E) were also the ones giving strong signal in the DGGE fingerprinting. Sequences typically found in fertilized plots (T, II, III) were not detected in any of the DGGE fingerprints for control plots.
Diversity Indices
The different indices calculated for diversity estimation are presented in Table 2. Ash treatment did not affect the diversity indices of clone libraries, and the depth-related change in population was not reflected in a change of mcr diversity (P > 0.1 for H’ and α).
Phylogenetic Analysis
Inferred amino acid sequences were used to assess the phylogenetic relationships between all detected restriction groups. Sequences were separated in two broad clusters, previously defined as Rice cluster-I [12] and Fen-cluster [10] (Fig. 4). Sequences representing the most frequent restriction groups in both upper layer (B and L) and deeper layer (E) all clustered closely together (>94% identity). They were related to sequences belonging to Rice cluster-I. Sequences unique or most abundant in fertilized plots (T, II, III) all grouped together (>96% identity) within Fen cluster. It contains a novel group of mcr sequences from uncultured methanogens retrieved from a boreal fen. One sequence represented by a small number of clones (X) clustered with sequences previously retrieved from rice roots and was closely related to known members of the order Methanosarcinales (90% identity).

Phylogenetic dendrogram representing the relationship between mcr amino acid sequences (250 aa long) retrieved from drained bog and other related methanogenic Archaea. Sequences obtained from bog libraries are designated in boldface. The capital letter or number in the sequence name corresponds to the RFLP pattern. The tree was constructed using FITCH distance matrix analysis. GenBank accession numbers are indicated for all sequences. Methanopyrus kandleri was used as outgroup. The scale bar represents 10% sequence divergence. Bootstrap values are shown for each node.
Discussion
Maximum potential CH4 production in upper anaerobic layers and decrease of the production with depth, as detected in the Pelso-Resula drained bog, is typical for boreal peatlands and has been observed in different studies [10, 20, 47]. However, the rates of potential methane production were lower than the values reported for an undisturbed fen in Eastern Finland, even though sampling was done at the same time of the growing season [10]. The low potential CH4 production suggests a lack of adequate substrate for methanogenesis. Drainage causes typical mire vegetation to change toward forest vegetation [25]. The Pelso-Resula bog has been drained by ditching, and small pines together with some poorly growing birches grow on the site, reflecting the transformation stage of the bog. In natural peatlands, sedges have been shown to transport carbon as exudates to layers of maximum root density [19, 42]. After drainage, sedges decline and are replaced by emerging trees. Primary production increases fast above ground, while the major rooting zone concentrates organic matter in the first 30 cm below the peat surface and less carbon is allocated to anaerobic layers [22, 23]. Additionally, with lowered water table, recently produced organic matter will be efficiently degraded in the increased aerobic layers, limiting potential substrate for methanogenesis to reach anaerobic layers of the peat (e.g., [43]). The composition of the methanogen community reflects the nutrient condition of the drained bog. Mcr sequences (B, L, E) dominating the libraries were all previously found to be characteristic for deeper layers of peatland [10]. They grouped phylogenetically with members of Rice cluster-I. Mcr sequences belonging to Rice cluster-I were originally retrieved from rice roots [12] and have recently been shown to originate from hydrogenotroph methanogens [41]. This indicates that the methanogens in the drained bog, corresponding to deep-layer community of natural peatlands, are utilizing CO2:H2 for methane production. The result is in accordance with studies that have shown hydrogenotrophy to be the major methanogenic pathways in the deeper layers of peatlands [17, 26]. Acetate, which typically originates from root exudates or as an end product of fermentation, is probably not an important precursor for methanogenesis in the drained bog.
Ash fertilization affected the peat by increasing the pH marginally in the submerged, anaerobic layers of the bog. Previous results have shown ash fertilization to have a liming effect, raising the pH and increasing the amount of nutrients (P, K, Ca, and trace elements) in the aerobic layers of peatlands [33, 39]. The little change in pH observed here did not have any major influence on the methanogen community. The three restriction groups dominating the different peat layers were the same in both control and fertilized sites. However, some groups of sequences (II, III, T) were only retrieved from the fertilized plots. Those sequences were closely related pylogenetically and all grouped within the same clad of the Fen cluster. This cluster represents a new group of mcr sequences recently retrieved from a natural boreal fen [10]. Even though the precursors used by members of the Fen cluster for methane production remain unknown, grouping of the sequences with members of Methanomicrobiales may suggest that those originate from hydrogenotrophs.
Methanogen community structure differed significantly as a function of peat depth. As shown by both DGGE and RFLP analyses, mcr sequences retrieved from upper layers were different from those originating from deeper layers. Depths-related change in methanogen community has earlier been shown in a natural fen and may indicate an adaptation of the methanogens to changes of physicochemical conditions with depth [9, 10].
In summary, methanogen mcr sequences dominating the drained bog were similar to sequences retrieved from the deep layers of a natural fen. The main sequences grouped within the Rice cluster-I, indicating the presence of hydrogenotroph methanogens in the bog. Even though ash fertilization increased the pH of the bog, potential methane production was not influenced by fertilization and the main groups of methanogens did not change. However, some less common sequences were found only in the fertilized plots and may indicate a gradual change of the community. They all grouped within the newly defined Fen-cluster and may originate from methanogens responding to physicochemical changes induced by fertilization.
References
M Aarne (Eds) (1998) Finnish Statistical Yearbook of Forestry Finnish Forest Research Institute Finland
J Aarnio S Kaunisto M Moilanen H Veijalaneinen (1997) Suometsien lannoitus (Fertilization on drained peatlands) K Mielikäinen M Riikilä (Eds) Kannattava Puuntuotanto (Profitability of Forestry) Kustannusosakeyhtiö Metsälehti 116–126
S Altschul T Madden A Schaffer J Zhang Z Zhang W Miller D Lipman (1997) ArticleTitleGapped BLAST and PSI-BLAST: a new generation of protein database search programs Nucleic Acids Res 25 3389–3402 Occurrence Handle9254694
ASM (2000) Environmental Change: Microbial Contributions, Microbial Solutions. American Society for Microbiology. http://www.asmusa.org/pasrc/reports.htm
J Earl G Hall RW Pickup DA Ritchie C Edwards (2003) ArticleTitleAnalysis of methanogen diversity in a hypereutrophic lake using PCR-RFLP analysis of mcr sequences Microb Ecol 46 270–278 Occurrence Handle10.1007/s00248-003-2003-x Occurrence Handle14708751
J Ellermann R Hedderich R Bocher RK Thauer (1988) ArticleTitleThe final step in methane formation. Investigations with highly purified methyl-CoM reductase (component C) from Methanobacterium thermoautotrophicum (strain Marburg) Eur J Biochem 172 669–677 Occurrence Handle10.1111/j.1432-1033.1988.tb13941.x Occurrence Handle3350018
RA Fisher AS Corber CB Williams (1943) ArticleTitleThe relation between the number of species and the number of individuals in a random sample of an animal population J Anim Ecol 12 42–58
H Fritze J Perkiomaki U Saarela R Katainen P Tikka K Yrjala M Karp J Haimi M Romantschuk (2000) ArticleTitleEffect of Cd-containing wood ash on the microflora of coniferous forest humus FEMS Microbiol Ecol 32 43–51 Occurrence Handle10.1016/S0168-6496(00)00007-6 Occurrence Handle10779618
PE Galand H Fritze K Yrjälä (2003) ArticleTitleMicrosite-dependent changes in methanogenic populations in a boreal oligotrophic fen Environ Microbiol 5 1133–1143 Occurrence Handle10.1046/j.1462-2920.2003.00520.x Occurrence Handle14641593
PE Galand S Saarnio H Fritze K Yrjala (2002) ArticleTitleDepth related diversity of methanogen Archaea in Finnish oligotrophic fen FEMS Microbiol Ecol 42 441–449 Occurrence Handle10.1016/S0168-6496(02)00381-1
IJ Good (1953) ArticleTitleThe population frequencies of species and the estimation of the population parameters Biometrika 40 237–264
R Grosskopf S Stubner W Liesack (1998) ArticleTitleNovel euryarchaeotal lineages detected on rice roots and in the anoxic bulk soil of flooded rice microcosms Appl Environ Microbiol 64 4983–4989 Occurrence Handle9835592
D Hagerberg H Wallander (2002) ArticleTitleThe impact of forest residue removal and wood ash amendment on the growth of the ectomycorrhizal external mycelium FEMS Microbiol Ecol 39 139–146 Occurrence Handle10.1016/S0168-6496(01)00207-0
BA Hales C Edwards DA Ritchie G Hall RW Pickup JR Saunders (1996) ArticleTitleIsolation and identification of methanogen-specific DNA from blanket bog peat by PCR amplification and sequence analysis Appl Environ Microbiol 62 668–675 Occurrence Handle8593069
Hammer, O, Harper, DAT (2002) Data analysis package. Distributed by the author; available from: http://folk.uio.no/ohammer/past/
D Higgins J Thompson T Gibson JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
MA Horn C Matthies K Kusel A Schramm HL Drake (2003) ArticleTitleHydrogenotrophic methanogenesis by moderately acid-tolerant methanogens of a methane-emitting acidic peat Appl Environ Microbiol 69 74–83 Occurrence Handle10.1128/AEM.69.1.74-83.2003 Occurrence Handle12513979
InstitutionalAuthorNameIPCC ( 2001) Climate Change 2001 (The Scientific Basis) Cambridge University Press Cambridge, UK
A Joabsson TR Christensen (2001) ArticleTitleMethane emissions from wetlands and their relationship with vascular plants: an Arctic example Global Change Biol 7 919–932 Occurrence Handle10.1046/j.1354-1013.2001.00044.x
A Kettunen V Kaitala A Lehtinen A Lohila J Alm J Silvola PJ Martikainen (1999) ArticleTitleMethane production and oxidation potentials in relation to water table fluctuations in two boreal mires Soil Biol Biochem 31 1741–1749 Occurrence Handle10.1016/S0038-0717(99)00093-0
VM Komulainen H Nykänen PJ Martikainen J Laine (1998) ArticleTitleShort-term effect of restoration on vegetation change and methane emissions from peatlands drained for forestry in southern Finland Can J For Res 28 402–411 Occurrence Handle10.1139/cjfr-28-3-402
R Laiho J Laine (1997) ArticleTitleTree stand biomass and carbon content in an age sequence of drained pine mires in southern Finland For Ecol Manage 93 161–169
R Laiho H Vasander T Penttila J Laine (2003) ArticleTitleDynamics of plant-mediated organic matter and nutrient cycling following water-level drawdown in boreal peatlands Gbobal Biogeochem Cycles 17 1–22
Laine, J, Vasander, H, (1996) Ecology and vegetation gradients of peatlands. In: Vasander, H (Ed.) Peatlands in Finland. Finnish Peatland Society, pp 10–19
J Laine H Vasander R Laiho (1995) ArticleTitleLong-term effects of water level drawdown on the vegetation of drained pine mires in southern Finland J Appl Ecol 32 785–802
JM Lansdown PD Quay SL King (1992) ArticleTitleCH4 production via CO2 reduction in a temperate bog: a source of 13C-depleted CH4 Geochim Cosmochim Acta 56 3493–3503 Occurrence Handle10.1016/0016-7037(92)90393-W
E Lappalainen (1996) Mires of Finland and their uses E Lappalainen (Eds) Global Peat Resources International Peat Society Jyskä 69–74
D Lloyd KL Thomas A Hayes B Hill BA Hales C Edwards JR Saunders DA Ritchie M Upton (1998) ArticleTitleMicro-ecology of peat: minimally invasive analysis using confocal laser scanning microscopy, membrane inlet mass spectrometry and PCR amplification of methanogen-specific gene sequences FEMS Microbiol Ecol 25 179–188 Occurrence Handle10.1016/S0168-6496(97)00094-9
T Lueders KJ Chin R Conrad M Friedrich (2001) ArticleTitleMolecular analyses of methyl-coenzyme M reductase alpha-subunit (mcrA) genes in rice field soil and enrichment cultures reveal the methanogenic phenotype of a novel archaeal lineage Environ Microbiol 3 194–204 Occurrence Handle10.1046/j.1462-2920.2001.00179.x Occurrence Handle11321536
PE Luton JM Wayne RJ Sharp PW Riley (2002) ArticleTitleThe mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill Microbiology 148 3521–3530 Occurrence Handle12427943
S Mahmood RD Finlay H Wallander S Erland (2002) ArticleTitleEctomycorrhizal colonisation of roots and ash granules in a spruce forest treated with granulated wood ash For Ecol Manage 160 1–3
PJ Martikainen H Nykaenen J Alm J Silvola (1995) ArticleTitleChange in fluxes of carbon dioxide, methane and nitrous oxide due to forest drainage of mire sites of different trophy Plant Soil 168 571–577
M Moilanen K Silfverberg TJ Hokkanen (2002) ArticleTitleEffects of wood-ash on the tree growth, vegetation and substrate quality of a drained mire: a case study Forest Ecol Manag 171 321–338 Occurrence Handle10.1016/S0378-1127(01)00789-7
C Moyer F Dobbs D Karl (1994) ArticleTitleEstimation of diversity and community structure through restriction fragment length polymorphism distribution analysis of bacterial 16S rRNA genes from a microbial mat at an active, hydrothermal vent system, Loihi Seamount, Hawaii Appl Environ Microbiol 60 871–879 Occurrence Handle7512808
G Muyzer T Brinkhoff U Nubel C Santegoeds H Schafer C Wawer (1998) Denaturing gradient gel electrophoresis (DGGE) in microbial ecology ADL Akkermans JD Elsas Particlevan FJ Bruijn Particlede (Eds) Molecular Microbial Ecology Manual Kluwer Academic Dordrecht 1–27
D Nercessian M Upton D Lloyd C Edwards (1999) ArticleTitlePhylogenetic analysis of peat bog methanogen populations FEMS Microbiol Ecol 173 425–429 Occurrence Handle10.1016/S0378-1097(99)00073-7
RM Niemi I Heiskanen K Wallenius K Lindstrom (2001) ArticleTitleExtraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia J Microbiol Methods 45 155–165 Occurrence Handle10.1016/S0167-7012(01)00253-6 Occurrence Handle11348673
E Paavilainen J Päivänen (1995) Peatland Forestry—Ecology and Principles Ecological Studies: Analysis and Synthesis, vol 111 Springer Berlin 248
A Saarsalmi E Malkonen (2001) ArticleTitleForest fertilization research in Finland: a literature review Scand J For Res 16 514–535
CE Shannon W Weaver (1963) The Mathematical Theory of Communication University of Illinois Press Urbana, IL
MV Sizova NS Panikov TP Tourova PW Flanagan (2003) ArticleTitleIsolation and characterization of oligotrophic acido-tolerant methanogenic consortia from a Sphagnum peat bog FEMS Microbiol Ecol 45 301–315 Occurrence Handle10.1016/S0168-6496(03)00165-X
L Strom A Ekberg M Mastepanov TB Christensen (2003) ArticleTitleThe effect of vascular plants on carbon turnover and methane emissions from a tundra wetland Global Change Biol 9 1185–1192 Occurrence Handle10.1046/j.1365-2486.2003.00655.x
BH Svensson I Sundh (1992) ArticleTitleFactors affecting methane production in peat soils Suo 43 183–190
RK Thauer (1998) ArticleTitleBiochemistry of methanogenesis: a tribute to Marjory Stephenson Microbiology 144 2377–2406 Occurrence Handle9782487
M Upton B Hill C Edwards JR Saunders DA Ritchie D Lloyd (2000) ArticleTitleCombined molecular ecological and confocal laser scanning microscopic analysis of peat bog methanogen populations FEMS Microbiol Lett 193 275–281 Occurrence Handle10.1016/S0378-1097(00)00496-1 Occurrence Handle11111036
CF Weil BA Sherf JN Reeve (1989) ArticleTitleA comparison of the methyl reductase genes and gene products Can J Microbiol 35 101–108 Occurrence Handle2720489
RT Williams RL Crawford (1984) ArticleTitleMethane production in Minnesota peatlands Appl Environ Microbiol 47 1266–1271
Acknowledgments
We thank Mikko Moilanen and Jorma Issakainen for taking samples. The work was funded by the Finnish Academy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Galand, P., Juottonen, H., Fritze, H. et al. Methanogen Communities in a Drained Bog: Effect of Ash Fertilization. Microb Ecol 49, 209–217 (2005). https://doi.org/10.1007/s00248-003-0229-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-003-0229-2