
Abstract
Effective surveillance is necessary for early detection of tumors in children with cancer predisposition syndromes. Instituting a surveillance regimen in children comes with practical challenges that include determining imaging modality and timing, and considering cost efficiency, accessibility, and the significant consequences of false-positive and false-negative results. To address these challenges, the American Association for Cancer Research has recently published consensus recommendations that focus on surveillance of cancer predisposition syndromes in children. This review condenses the imaging surveillance recommendations for syndromes that carry a predisposition to renal tumors in childhood, and includes summaries of the predisposition syndromes and discussion of considerations of available imaging modalities.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is fundamentally a genetic disease, resulting from pathogenic mutations that act through complex mechanisms to produce dysregulation of the core evolutionary processes of cellular proliferation, differentiation and survival. The genetic underpinnings of cancer were first deduced in the 19th and early 20th centuries by Paul Broca, Theodor Boveri and Aldred Warthin, by study of the incidence of similar cancers among related individuals [1]. Although we now know that most cancers result from somatic (also called post-zygotic, sporadic or acquired) mutations, the study of cancers caused by familial predisposition — germline mutations — has greatly contributed to the understanding of cancer genetics. One illustrative example is Alfred Knudson’s “two-hit” hypothesis, derived from his analysis of the epidemiology of hereditary retinoblastoma. This postulates that if one mutation is inherited in the germline, a second mutation has to be acquired to produce the cancer. This hypothesis was corroborated with the discovery of the retinoblastoma predisposition gene, RB1, and it has since been a guiding principle in the study of cancer pathogenesis [1, 2].
Cancer predisposition genes
Cancer predisposition gene mutations occur in the germline and can be categorized as either tumor-suppressor or oncogenes. Mutations in the former result in proteins whose loss of function then enables the onset of cancer through abrogation of their regulatory role in inhibiting cell cycle progression, promoting apoptosis, or stimulating differentiation. Mutations in the latter — genes that often code for kinases — produce a gain of function, resulting in a constitutively active protein that results in proliferation. The majority of cancer predisposition genes are tumor suppressors [3].
Specific types and sites of mutations in cancer predisposition genes are often associated with a specific cancer susceptibility. Different mutations can also confer different risks of the same cancer, as well as different risks of different cancers. For example, SMARCB1 mutations can predispose a child to either renal rhabdoid tumor or schwannomatosis and meningioma, based on the type of mutation [4, 5]. How particular variations in the same gene produce such strikingly different phenotypes remains a conundrum in cancer genetics, indicating uncharted complexities of gene function. These are influenced by mosaicism and organ-specificity, and also by intricate interactions in haplotypic background and between epigenetic and non-genetic factors. The fact that some cancer predisposition syndromes have a defined cancer spectrum (e.g., hereditary retinoblastoma and Beckwith–Wiedemann syndrome), whereas others have a variable cancer spectrum (e.g., Li–Fraumeni syndrome and Von Hippel–Lindau syndrome) is evidence of these complex interactions.
In children, it is estimated that at least 10% of cancer patients harbor a germline mutation in a cancer predisposition gene, and this number is likely an underestimate because of undiscovered genes, variants and syndromes [6]. In children, genetic predisposition is suggested when any of the following occur: (a) a family history of related cancers, (b) physical findings suggestive of a cancer predisposition syndrome, (c) multifocal or multiple cancers, (d) the onset of cancer earlier than is typical for similar sporadic tumors or (e) the onset of tumors specific for a cancer predisposition syndrome (e.g., adrenocortical carcinoma in Li–Fraumeni syndrome) [2, 3].
Surveillance for cancer predisposition syndromes
A child suspected of having cancer predisposition should undergo genetic testing to confirm predisposition and characterize the pathogenic variant. The determination of cancer predisposition provides prognostic information that can be used to establish a surveillance regimen, which can include imaging, physical examination and blood tests for biomarkers. Given that a child is predisposed to cancer, it is logical that surveillance should provide earlier detection of cancer, and that this should result in lower morbidity from less intensive therapy and higher rates of survival. Although there is evidence in support of surveillance, robust supporting evidence on outcomes has not been established, largely because of the low incidence of known cancer predisposition syndromes. An early study on outcomes of children with Beckwith–Wiedemann syndrome found that screening protocols with ultrasonography (US) were associated with detection of smaller tumors [7].
The diagnosis of a cancer predisposition syndrome influences management by enabling pre-symptomatic testing of relatives and counseling for family planning. Once a tumor is detected, information from genetic testing can be used to target vulnerabilities specific to the tumor type. For example, such data can determine feasibility of specific upfront chemotherapy or partial nephrectomy. This trend toward personalized cancer therapy is only beginning [1].
Effective surveillance must be cost-effective, adequately accurate in detection, and appropriately low-risk. Practical challenges with cancer surveillance in children include the choice of modality, timing (i.e. when to start, when to stop, intervals between surveillance tests), and whether tests should change over time [2, 3]. The cost incurred by families undergoing imaging surveillance, as well as anxiety associated with scanning, can be significant. False-negative results can have significant consequences, and false-positives can result in unnecessary intervention and associated psychological distress, morbidity and cost.
To address these challenges, the Pediatric Cancer Working Group of the American Association for Cancer Research conducted a workshop in 2016 to develop consensus recommendations for surveillance in the most common cancer predisposition syndromes. An estimated childhood cancer risk of ≥5% was set as the threshold to always recommend screening, and screening was sometimes recommended for syndromes with estimated risk 1–5%. The recommendations would not apply to follow-up of diagnosed tumors or surveillance for secondary malignant neoplasms, and any positive finding or new clinical signs would warrant vigilant imaging follow-up or intervention. These recommendations were presented in the summer of 2017 in a series of articles in Clinical Cancer Research. This review condenses the imaging recommendations from this series and focuses on syndromes with predisposition to renal tumors.
Imaging modalities
Imaging surveillance is suited for solid tumors, where outcomes are more clearly associated with the stage of the tumor at diagnosis. Whether to include imaging in the surveillance protocol, and the type of modality, is determined by the purported sensitivity and specificity of the modality, cost-effectiveness, the use of ionizing radiation, and ease of performance. Accordingly, current surveillance protocols largely rely on MRI (both whole-body and organ-specific) and US. These studies should be interpreted by a radiologist with experience in cancer predisposition syndromes [8]. Efficient reporting of imaging results and open communication with patient families about tumor risk are important in reducing anxiety and aiding compliance with surveillance [9].
Ultrasound
Although outcomes data on US for surveillance are limited, McNeil et al.[10] demonstrated cost-effectiveness and survival benefit for people with Beckwith–Wiedemann syndrome who were screened for Wilms tumor and hepatoblastoma. Advantages of US include ease of use and lack of ionizing radiation. Scans require less time than MRI, and sedation is rarely necessary. Spatial resolution in small children is excellent. However, contrast resolution is limited, as is spatial resolution in larger patients.
Performance of US is operator-dependent, but with appropriate technique and patient factors, US can have similar accuracy to CT for renal tumors [11]. The detection of smaller lesions requires sonographer vigilance and experience, sometimes with repeat scanning by the radiologist. US should be performed per American College of Radiology practice parameters, should utilize a probe appropriate for patient size and the tissues being imaged, and should include gray-scale and Doppler imaging. The use of cine clips is also beneficial. Contrast-enhanced ultrasound has not been validated for surveillance but can be used for lesion characterization. Ultrasound contrast agents (fluoride molecular gas in minuscule lipid or protein shells) are well-tolerated but administration requires intravenous access.
Magnetic resonance imaging
Depending on the cancer predisposition syndromes, MRI surveillance for renal tumors might be performed with whole-body scanning or targeted abdominal or renal MRI. Whole-body MRI consists of contiguous multi-region scanning to produce head-to-toe imaging. The capacity for whole-body MRI to detect lesions depends on several factors, including the anatomical site, size and lesion histology [12]. Whole-body MRI has shown good diagnostic accuracy for staging solid tumors. There is no robust evidence of outcomes of whole-body MRI surveillance, but several recent studies support its use for Li–Fraumeni syndrome, hereditary pheochromocytoma/paraganglioma syndrome, and constitutional mismatch repair deficiency [13,14,15]. Whole-body MRI has been shown to have superior sensitivity and similar specificity compared to biochemical surveillance for detecting paraganglioma in hereditary pheochromocytoma/paraganglioma syndrome, and whole-body MRI surveillance has also been found to be beneficial in Von Hippel–Lindau syndrome [16, 17].
Renal protocol MRI typically consists of axial fast spin-echo T2-weighted and axial gradient T1-weighted in- and out-of-phase sequences. Intravenous contrast agent is necessary to characterize enhancement of lesions, and macrocyclic agents potentially afford decreased risk of nephrogenic systemic fibrosis. The contrast enhanced portion often consists of axial or coronal pre-contrast, followed by fast gradient echo T1 fat-saturated images obtained at 30 s, 90–100 s, 180–210 s and 5–7 min. Diffusion-weighted imaging is performed in axial plane with b values of 0–50 s/mm2, 400–500 s/mm2 and 800–1,000 s/mm2.
Wilms tumor predisposition syndromes
Wilms tumor predisposition states can be subdivided into overgrowth syndromes and other Wilms tumor-related syndromes. Wilms tumor risk, median age at presentation, and incidence for the various syndromes are given in Table 1. Imaging surveillance recommendations for these syndromes are directed toward early detection and are presented in Table 2.
Overgrowth syndromes associated with Wilms tumor
These syndromes might confer predisposition to Wilms tumor and hepatoblastoma. Overall predisposition syndromes account for approximately 20% of Wilms tumor, but at least 80% of hepatoblastomas are associated with a predisposition syndrome [18, 19].
Beckwith–Wiedemann syndrome
Beckwith–Wiedemann syndrome stands as the prototype predisposition syndrome for Wilms tumor (Fig. 1). It affects at least 1 in 11,000 children and is characterized by overgrowth, macroglossia, omphalocele and hemihyperplasia, with predisposition for Wilms tumor and hepatoblastoma. Clinical features are attributed to genetic and epigenetic changes in chromosome 11p15. Paternal disomy of 11 (UPD11) and methylation at imprinting center 1 (IC1) confer a high risk of tumor (up to about 30%) [20, 21]. Low-risk (epi)genotypes include CDKN1C variants and loss of methylation at IC2. The former also confers a neuroblastoma predisposition. Idiopathic hemihyperplasia does not have additional features of Beckwith–Wiedemann syndrome but carries an elevated risk of Wilms tumor. It is thought to be a forme fruste of an “11p overgrowth” spectrum.

Beckwith–Wiedemann syndrome. Annual renal/bladder imaging follow-up in a 6-year-old boy with left kidney hydronephrosis evident since prenatal imaging. a, b Axial (a) and coronal (b) magnetic resonance urography images show a 1-cm lesion that is hypoechoic to renal cortex (arrows). Nephron-sparing surgery revealed histopathology with a mixture of nephrogenic rest and Wilms tumor without a capsule, indicative of an evolving Wilms tumor. Genetic consult identified macroglossia and hemihypertrophy and genetic testing showed hypermethylation of IC1, consistent with Beckwith–Wiedemann syndrome
Current surveillance recommendations in the United States do not differentiate high- from low-risk genotypes for Beckwith–Wiedemann syndrome. Recommendations are instituted based on estimated risk of cancer incidence of at least 1% and these are likely to evolve to incorporate genotypic distinctions as they are further characterized. Based on an epidemiologic analysis, imaging surveillance recommendations for Wilms tumor consist of renal US every 3 months through the 7th birthday; imaging surveillance for hepatoblastoma adds abdominal US (which can supplant the renal US) every 3 months through the 4th birthday, with monitoring of alpha fetoprotein (AFP) levels. Abdominal MRI is recommended for further characterization of any lesion seen on US, or if serum AFP increases over two successive tests 6 weeks apart [19].
Simpson–Golabi–Behmel syndrome
Simpson–Golabi–Behmel syndrome is characterized by macrosomia, distinctive facies and intellectual disability, as well as genitourinary and gastrointestinal anomalies. Skeletal anomalies include segmentation abnormalities of the ribs and spine, and congenital hip dislocation. Mutations are in GPC3 or GP4 genes. Commonest associated tumors are Wilms tumor and liver tumors. Recommendation for tumor screening is similar to that for Beckwith–Wiedemann syndrome, with screening for Wilms tumor and hepatoblastoma.
Perelman syndrome
Perelman syndrome, caused by mutations in DIS3L2, is a rare overgrowth syndrome characterized by macrosomia, hypotonia, distinctive facies, visceromegaly, renal dysplasia and nephroblastomatosis (Fig. 2) [19, 22]. Most children have early mortality but survivors are at high risk for early and multifocal Wilms tumor. Perelman syndrome is notable for polyhydramnios and fetal ascites and the absence of macroglossia and omphalocele. Tumor surveillance is recommended only for Wilms tumor.

Nephroblastomatosis in a baby boy with Perelman syndrome with deletion at 2q37.1, including the DIS3L2 locus. a, b Longitudinal left kidney (a) and transverse right kidney (b) US images at 5 months of age show diffusely enlarged kidneys with peripheral nodules. Dominant nodules are marked with arrows, and the large nodule in the right lower pole is mildly hyperechoic. c MRI was obtained for further characterization of masses. Coronal inversion recovery sequence shows uniform mild T2 prolongation in all the masses (arrowheads). d Apparent diffusion coefficient (ADC) map from diffusion-weighted MR sequence shows restricted diffusion (dark areas) in the nephrogenic rests. Lesions were responsive to chemotherapy with vincristine and dactinomycin
Other Wilms tumor predisposition syndromes
WAGR syndrome
WAGR syndrome (Wilms tumor, aniridia, genitourinary abnormalities and retardation) is caused by deletions in chromosome 11p13, involving WT1 and PAX6. The product of WT1 is a transcription factor that regulates differentiation and growth of the kidneys, gonads, spleen and mesothelium. Nephropathy might also be a feature. The risk of Wilms tumor is approximately 50%.
Denys–Drash and Fraser syndromes
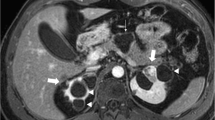
Denys-Drash syndrome is characterized by Wilms tumor, nephrotic syndrome and gonadal dysgenesis with a 46XY karyotype, is also caused by pathogenic variants of WT1 (Fig. 3). Fraser syndrome shares similar features and is characterized by Wilms tumor, proteinuria from focal segmental glomerulosclerosis, and gonadal dysgenesis with a 46XY karyotype. There is also a risk for gonadoblastoma. Denys–Drash and Fraser syndromes might be part of the same spectrum of disease. The risk of Wilms tumor is more than 90% and could be related to truncating mutations in exon 8/9 of WT1. Wilms tumor occurs earlier in children with WAGR, Denys–Drash and Fraser syndromes than in those with somatic WT1 mutations.

Denys–Drash syndrome in a 10-month-old girl who presented with hypertension and bilateral renal masses on renal US. a Right renal US shows a fairly homogeneous and mildly hyperechoic mass, 16 cm in diameter. b Left renal US shows a heterogeneous, echogenic 3-cm mass with small cystic areas. c Coronal T2-weighted MR image shows a large heterogeneous mass in the right kidney (arrows) and multi-cystic foci in the upper and lower poles of the left kidney (arrowheads). d Diffusion-weighted MR imaging apparent diffusion coefficient (ADC) map shows restricted diffusion (arrows) with central necrotic areas of T2 shine-through in the right kidney and cystic changes of T2 shine-through with dilated glomeruli in the left kidney (arrowheads) as well as focal areas of restricted diffusion. Note that the diffusion-weighted image enhances the detection the nephrogenic rest. Pathology after right nephrectomy showed Wilms tumor with stromal-predominant histology. Left renal biopsy revealed intralobar nephrogenic rest with cyst formation. During chemotherapy, the girl developed nephrotic syndrome. Germline blood testing revealed a WT1 mutation. She was diagnosed with Denys–Drash syndrome
Bohring–Opitz syndrome and others
Bohring–Opitz syndrome is rare and is characterized by developmental delay, distinctive facies, radial head dislocation with ulnar deviation, and brain deformity. The syndrome is associated with ASXL1 mutations, early mortality and increased Wilms tumor risk. Mulibrey (“muscle, liver, brain”) nanism is also a rare syndrome producing growth retardation, hepatomegaly, distinct dysmorphism and constrictive pericarditis. The largest known cohort originates from Finland, and it is caused by mutations in TRIM37. A surveillance regimen similar to that for Beckwith–Wiedemann syndrome is recommended for WAGR syndrome, Denys–Drash syndrome, Fraser syndrome, Bohring–Opitz syndrome and mulibrey nanism. Last, it should be noted that MYCN copy number gains in the germline have recently been shown to predispose to Wilms tumor [23].
Trisomy 18
Trisomy 18 is the second-most common trisomy and is marked by a variety of malformations and severe developmental delay, and poor survival. There is an increased risk of cancer, most commonly Wilms tumor and hepatoblastoma. Cancer screening for trisomy 18 is controversial, but screening recommendations are similar to those for Beckwith–Wiedemann syndrome [19].
DICER1 syndrome
DICER1 codes for a ribonuclease III that processes micro-ribonucleic acid (miRNA), crucial for activating RNA interference, a process that limits gene expression. Pleuropulmonary blastoma is the most common childhood lung tumor and it is associated with DICER1 syndrome. Other DICER1-related tumors are embryonal rhabdomyosarcoma of the cervix, ovarian Sertoli–Leydig cell tumor and gynandroblastoma. Multinodular goiter is common in DICER1 syndrome, and there is elevated risk for differentiated thyroid carcinoma. Multilocular cystic nephroma is also common in children with DICER1 syndrome. Interestingly, rare mosaic variants of a particular domain of the ribonuclease result in a Wilms tumor overgrowth syndrome known as GLOW syndrome, which presents with global developmental delay, lung cysts, overgrowth and Wilms tumor. Wilms tumor risk in DICER1 syndrome extends to a later age than with Wilms tumor-related cancer predisposition syndromes described above. Rare brain tumors, most commonly pineoblastoma and pituitary blastoma, have been reported in DICER1 syndrome.
For renal tumors, particularly multilocular cystic nephroma and Wilms tumor, abdominal US is recommended, albeit at a different schedule than for other Wilms tumor predisposition syndromes — biannual abdominal US until age 8 and annually after 8 years. Given the wide variety of tumors with this syndrome, some institutions use whole-body MRI for surveillance in children older than 6 years [24].
Hyperparathyroid–jaw tumor syndrome
This syndrome is characterized by parathyroid adenoma and ossifying maxillary or mandibular fibroma, with elevated risk for parathyroid carcinoma. Other features include uterine tumors, renal cysts and other anomalies, and rarely Wilms tumor. Hyperparathyroid–jaw tumor syndrome is caused by pathogenic variants in CDC73, which codes for parafibromin, a tumor suppressor probably involved in regulating transcription and cell cycle progression.
Surveillance should begin between 5 years and 10 years of age and include dental and mandibular radiographs and renal ultrasound every 5 years, in addition to annual serum calcium panel for hyperparathyroidism. Pelvic US is reserved for females with symptoms (e.g., abnormal bleeding) [25].
Li–Fraumeni syndrome
Li–Fraumeni syndrome results in a wide spectrum of tumors, caused by mutations in the suppressor gene TP53. Children with Li–Fraumeni syndrome might develop tumors of the kidney, including Wilms tumor [8, 26]. During the first 18 years of life, people with Li–Fraumeni syndrome should undergo annual surveillance with whole-body MRI and brain MRI. Additionally, abdominal and pelvic US are performed every 3–4 months, as well as vigilant clinical examinations [8].
Renal cell carcinoma predisposition syndromes
Body imaging surveillance recommendations for these syndromes are given in Table 3.
PTEN tumor syndrome
PTEN (phosphatase and tensin homolog) tumor syndrome consists of associated tumor syndromes including Cowden, Proteus and Bannayan–Riley–Ruvalcaba syndromes. Shared clinical features include macrocephaly, gastrointestinal polyposis, lipomata, intellectual disability and vascular malformations. The PTEN gene is a tumor suppressor that codes for the phosphatase and tensin homolog and negatively regulates the PI3K/AKT/mTOR pathway, which plays a complex and crucial role in in cell proliferation and angiogenesis. PTEN tumor syndrome confers an elevated risk of melanoma, breast, thyroid, endometrial, colorectal and renal cell carcinoma, usually the clear cell subtype. Thyroid carcinoma (papillary and follicular) is the predominant childhood cancer. Currently there are no screening recommendations specific to renal cancer risk in the pediatric population [24].
Hereditary leiomyomatosis and renal cell cancer syndrome
Hereditary leiomyomatosis and renal cell cancer syndrome is caused by pathogenic variants in the fumarate hydratase gene FH, a component of the Krebs cycle. The build-up of fumarate is thought to activate hypoxia-inducible factor, which regulates gene expression by oxygen and is implicated in promoting tumor growth, angiogenesis and metastasis. It is characterized by painful cutaneous and uterine leiomyomata and renal cell carcinoma. Leiomyomata occur earlier and are more severe than is typical, and rarely transform to leiomyosarcoma. Certain pathogenic variants are also associated with pheochromocytoma and paraganglioma.
Renal cell carcinoma are papillary type 2 and aggressive; small lesions metastasize early. Early detection might improve morbidity and mortality. Because renal cell carcinoma in hereditary leiomyomatosis and renal cell cancer syndrome tends to be small and isoechoic, annual renal protocol MRI beginning at age 8 (including chemical shift imaging, at least three contrast phases of dynamic 3-D gradient T1-weighted imaging and diffusion-weighted imaging [DWI]) is favored over US. Screening for leiomyomata is suggested with gynecologic US starting at age 20 [24].
Hereditary pheochromocytoma/paraganglioma syndrome
This syndrome predisposes to tumors of neural crest origin, pheochromocytoma and paraganglioma. These are located in the paravertebral regions, from the skull to the pelvis. Other tumors that might develop are clear cell renal cell carcinoma, pituitary adenoma and gastrointestinal stromal tumor of the stomach.
The genes most commonly responsible are of the succinate dehydrogenase enzyme complex, (SDHx), which participates in the Krebs cycle and the electron transport chain. Elevated succinate concentrations prevent degradation of hypoxia-inducible factor, which is implicated in tumor promotion, metastasis and angiogenesis. A higher risk of malignancy has been recognized for SDHB mutations. Despite the genotype-phenotype association with SDHB, distinct surveillance paradigms based on genotype are not currently recommended. Given anatomical distribution of paragangliomata, biannual whole-body MRI is recommended, beginning at age 6–8 years. Concomitant neck MRI with contrast might be considered [27].
Von Hippel–Lindau syndrome
Von Hippel–Lindau syndrome predisposes to renal and pancreatic cysts and multiple tumors, including hemangioblastoma (cerebellar, brainstem, spinal and retinal), clear cell renal cell carcinoma, pheochromocytoma, pancreatic neuroendocrine tumor, endolymphatic sac tumor, ovarian broad ligament and epididymal cystadenoma. Pediatric patients are particularly at risk for hemangioblastoma and pheochromocytoma. The incidence of Von Hippel–Lindau syndrome is relatively high for a cancer predisposition syndrome, at one in 36,000. The protein encoded by VHL is involved in the ubiquitination and degradation of hypoxia-inducible factor, a pathogenic actor also involved in hereditary leiomyomatosis and renal cell cancer and hereditary pheochromocytoma/paraganglioma syndromes. There are four types of Von Hippel–Lindau syndrome, with varying risks of different tumors and phenotype defined by the genotype. A relatively high risk of renal cell carcinoma is conferred by truncating mutations and exon deletions (type 1 Von Hippel–Lindau syndrome). Higher hypoxia-inducible factor expression appears to result in lower risk for renal cell carcinoma.
Everyone with Von Hippel–Lindau syndrome develops a tumor by age 75. Lifelong surveillance is therefore necessary. Imaging surveillance of the central nervous system begins at age 8 with biannual contrast-enhanced brain MRI with high-resolution imaging of the internal auditory canals (for risk of endolymphatic sac tumor) and biannual spine MRI with contrast. For body tumors, annual MRI of the abdomen with renal protocol is recommended beginning at age 10 and can be performed concurrently with the brain and spine MRI. The remainder of Von Hippel–Lindau syndrome surveillance is clinical, including screening for pheochromocytoma with plasma-free metanephrines or 24-h urine fractionated metanephrines [27]. Small screening-detected tumors undergo active surveillance if less than 3 cm in diameter [28]. Tumor growth reaching 3 cm or more in diameter undergoes a nephron-sparing surgery, or a partial nephrectomy or percutaneous thermal ablation as an alternative, depending on lesion size [26].
It should be noted that an increased risk of clear cell renal cell carcinoma is also seen in people with translocations of chromosome 3p, which can involve various genes including VHL, SETD2, PBRM1 and with inactivation of germline BRCA-associated protein 1 gene (BAP1) [29, 30]. For individuals with a germline mutation of BAP1, the information on the most appropriate screening modality is limited.
Other renal tumor predisposition syndromes
Constitutional mismatch repair deficiency
The mismatch repair system is responsible for recognizing and repairing errors during deoxyribonucleic acid (DNA) replication and DNA damage. Mismatch repair genes include MSH2, MSH6, MLH1 and PMS2. Biallelic germline mutations of these genes result in constitutional mismatch repair deficiency syndrome in children. In contrast, inherited heterozygous mutations in these genes result in Lynch syndrome, associated with adult colorectal and ovarian/endometrial cancers.
The experience with solid tumors during childhood in Lynch syndrome is scant, but the pediatric tumor spectrum of constitutional mismatch repair deficiency syndrome is wide and includes renal lesions. There is a significant risk (about 10%) of upper tract urothelial cancers [31, 32], especially in those with MSH2 and MLH1 mutations [33]. Surveillance protocols focus on the different tumor frequencies at specific ages, and surveillance of the genitourinary tract is usually reserved for older children [34,35,36]. Recently, whole-body MRI has been added as a surveillance modality for constitutional mismatch repair deficiency syndrome, annually, starting at 6 years, or earlier if anesthesia is not necessary. Brain MRI is performed every 6 months to detect central nervous system lesions, while upper endoscopy and video capsule endoscopy start from age 8 [37].
Rhabdoid tumor predisposition syndrome
Rhabdoid tumors occur most often in the central nervous system (atypical teratoid/rhabdoid tumor) or kidney, and despite their name, they consist of undifferentiated small round blue cells with epithelial and mesenchymal components. Tumors are aggressive and present between ages 1 and 3. Rhabdoid tumor predisposition syndrome is characterized by loss-of-function germline mutation in SMARCB1 for rhabdoid tumor predisposition syndrome type 1, and SMARCA4 for rhabdoid tumor predisposition syndrome type 2. These gene products are involved in chromatin remodeling. This family of genes is also implicated in neurofibromatosis type 2-related disorders. SMARCB1 truncating mutation carriers can also develop schwannomatosis, multiple meningiomata, and malignant peripheral nerve sheath tumors. SMARCA4 mutations are strongly associated with renal rhabdoid tumor and small cell ovarian cancer hyporcalcemic type (which is similar on pathological analysis to rhabdoid tumor), but mutations show less penetrance in carriers [4].
Renal rhabdoid tumor is a highly aggressive malignancy. About 60% occur in the first year, with peak incidence between 6 months and 12 months. Tumors are centrally located and lobulated, with characteristic subcapsular fluid collections. Necrosis and calcification are common. Tumors quickly invade surrounding tissues and vessels, resulting in lung metastases [28].
Developing surveillance recommendations for rhabdoid tumor predisposition syndrome is complicated by the young age at onset, the aggressive nature of rhabdoid tumors, unclear penetrance and the rarity of the syndrome. Surveillance recommendations are specific to genotype. For SMARCB1 truncating mutation carriers, brain MRI every 3 months until age 5 is recommended. For surveillance of renal and extra-renal abdominal tumors, abdominal US every 3 months is recommended from infancy through age 5. One might consider whole-body MRI, to age 5 years, but there is not yet a recommended frequency for surveillance [4].
Conclusion
This review provides a summary of current imaging surveillance recommendations for cancer predisposition states with elevated risk for renal tumors. Surveillance is recognized as a means to improve survival in this vulnerable population, and imaging is a crucial part of surveillance for many syndromes. New syndromes, as well as more specific information about genotype–phenotype associations — perhaps revelations of cooperating epigenetic, genetic and environmental events that modify the phenotype conferred by the primary gene mutation — and outcomes data are expected to allow recommendations to be further refined, to be more tumor-specific and patient-specific. In addition, new techniques in molecular biology and advancements in diagnostic imaging are expected to provide less costly and more accurate methods of early cancer detection. Other risk-reducing measures, including preventative therapies, might also impact the demand for cancer surveillance.
References
Rahman N (2014) Realizing the promise of cancer predisposition genes. Nature 505:302–308
Brodeur GM, Nichols KE, Plon SE et al (2017) Pediatric cancer predisposition and surveillance: an overview, and a tribute to Alfred G. Knudson Jr. Clin Cancer Res 23:e1–e5
McGee RB, Nichols KE (2016) Introduction to cancer genetic susceptibility syndromes. Hematology Am Soc Hematol Educ Program 2016:293–301
Foulkes WD, Kamihara J, Evans DGR et al (2017) Cancer surveillance in Gorlin syndrome and rhabdoid tumor predisposition syndrome. Clin Cancer Res 23:e62–e67
Evans DGR, Salvador H, Chang VY et al (2017) Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 2 and related disorders. Clin Cancer Res 23:e54–e61
Zhang J, Walsh MF, Wu G et al (2015) Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373:2336–2346
Porteus MH, Narkool P, Neuberg D et al (2000) Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms' tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 18:2026–2031
Kratz CP, Achatz MI, Brugieres L et al (2017) Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res 23:e38–e45
Duffy KA, Grand KL, Zelley K, Kalish JM (2018) Tumor screening in Beckwith-Wiedemann syndrome: parental perspectives. J Genet Couns 27:844–853
McNeil DE, Brown M, Ching A, DeBaun MR (2001) Screening for Wilms tumor and hepatoblastoma in children with Beckwith-Wiedemann syndromes: a cost-effective model. Med Pediatr Oncol 37:349–356
Peng Y, Jia L, Sun N et al (2010) Assessment of cystic renal masses in children: comparison of multislice computed tomography and ultrasound imaging using the Bosniak classification system. Eur J Radiol 75:287–292
Guimaraes MD, Noschang J, Teixeira SR et al (2017) Whole-body MRI in pediatric patients with cancer. Cancer Imaging 17(6)
Malkin D (2014) Surveillance for children at genetic risk for cancer: are we ready? Pediatr Blood Cancer 61:1337–1338
Darge K, Jaramillo D, Siegel MJ (2008) Whole-body MRI in children: current status and future applications. Eur J Radiol 68:289–298
Anupindi SA, Bedoya MA, Lindell RB et al (2015) Diagnostic performance of whole-body MRI as a tool for cancer screening in children with genetic cancer-predisposing conditions. AJR Am J Roentgenol 205:400–408
Priesemann M, Davies KM, Perry LA et al (2006) Benefits of screening in von Hippel-Lindau disease — comparison of morbidity associated with initial tumours in affected parents and children. Horm Res 66:1–5
Jasperson KW, Kohlmann W, Gammon A et al (2014) Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Familial Cancer 13:257–265
Achatz MI, Porter CC, Brugieres L et al (2017) Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res 23:e107–e114
Kalish JM, Doros L, Helman LJ et al (2017) Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res 23:e115–e122
Maas SM, Vansenne F, Kadouch DJ et al (2016) Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A 170:2248–2260
Kalish JM, Deardorff MA (2016) Tumor screening in Beckwith-Wiedemann syndrome — to screen or not to screen? Am J Med Genet A 170:2261–2264
Fahmy J, Kaminsky CK, Parisi MT (1998) Perlman syndrome: a case report emphasizing its similarity to and distinction from Beckwith-Wiedemann and prune-belly syndromes. Pediatr Radiol 28:179–182
Williams RD, Chagtai T, Alcaide-German M et al (2015) Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6:7232–7243
Schultz KAP, Rednam SP, Kamihara J et al (2017) PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e76–e82
Wasserman JD, Tomlinson GE, Druker H et al (2017) Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e123–e132
Hartley AL, Birch JM, Tricker K et al (1993) Wilms' tumor in the Li-Fraumeni cancer family syndrome. Cancer Genet Cytogenet 67:133–135
Rednam SP, Erez A, Druker H et al (2017) Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: clinical features, genetics, and surveillance recommendations in childhood. Clin Cancer Res 23:e68–e75
Chung EM, Graeber AR, Conran RM (2016) Renal tumors of childhood: radiologic-pathologic correlation part 1. The 1st decade: from the radiologic pathology archives. Radiographics 36:499–522
Cancer Genome Atlas Research Network (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499:43–49
Farley MN, Schmidt LS, Mester JL et al (2013) A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 11:1061–1071
Mork M, Hubosky SG, Roupret M et al (2015) Lynch syndrome: a primer for urologists and panel recommendations. J Urol 194:21–29
Sijmons RH, Kiemeney LA, Witjes JA, Vasen HF (1998) Urinary tract cancer and hereditary nonpolyposis colorectal cancer: risks and screening options. J Urol 160:466–470
Barrow PJ, Ingham S, O'Hara C et al (2013) The spectrum of urological malignancy in lynch syndrome. Familial Cancer 12:57–63
Bakry D, Aronson M, Durno C et al (2014) Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer 50:987–996
Vasen HF, Ghorbanoghli Z, Bourdeaut F et al (2014) Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European consortium "care for CMMR-D" (C4CMMR-D). J Med Genet 51:283–293
Durno CA, Aronson M, Tabori U et al (2012) Oncologic surveillance for subjects with biallelic mismatch repair gene mutations: 10 year follow-up of a kindred. Pediatr Blood Cancer 59:652–656
Tabori U, Hansford JR, Achatz MI et al (2017) Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res 23:e32–e37
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Srinivasan, A.S., Saade-Lemus, S., Servaes, S.E. et al. Imaging surveillance for children with predisposition to renal tumors. Pediatr Radiol 49, 1453–1462 (2019). https://doi.org/10.1007/s00247-019-04432-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-019-04432-2