
Abstract
Background
DICER1 syndrome, arising from a mutation in the DICER1 gene mapped to chromosome 14q32, is associated with an increased risk of a range of benign and malignant neoplasms.
Objective
To determine the spectrum of abnormalities and imaging characteristics in patients with DICER1 syndrome at a tertiary pediatric hospital.
Materials and methods
This retrospective analysis evaluated imaging in patients ≤18 years with DICER1 germline variants between January 2004 and July 2016. An imaging database search including keywords pleuropulmonary blastoma, cystic nephroma, pineoblastoma, embryonal rhabdomyosarcoma, ovarian sex cord-stromal tumor, ovarian Sertoli-Leydig cell tumor and DICER1 syndrome, was cross-referenced against the institutional Cancer Genetics Program database, excluding patients with negative/unknown DICER1 gene testing.
Results
Sixteen patients were included (12 females; mean age at presentation: 4.2 years, range: 14 days to 17 years), with surveillance imaging encompassing the following modalities: chest X-ray and CT; abdominal, pelvic and neck US; and brain and whole-body MRI. Malignant lesions (68.8% of patients) included pleuropulmonary blastoma (5), pineoblastoma (3), ovarian Sertoli-Leydig cell tumor (1), embryonal rhabdomyosarcoma (1) and renal sarcoma (1); benign lesions (37.5% of patients) included thyroid cysts (2), thyroid nodules (2), cystic nephroma (2), renal cysts (1) and pineal cyst (1). A common lesional appearance observed across modalities and organs was defined as the “cracked windshield” sign.
Conclusion
The spectrum of DICER1-related tumors and the young age at presentation suggest early surveillance of at-risk patients is critical, while minimizing exposure to ionizing radiation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
DICER1 syndrome (OMIM#601200) is a familial cancer predisposition syndrome associated with inherited or de novo mutations in the DICER1 gene [1]. DICER1 is a gene located on chromosome 14q32.13 belonging to the ribonuclease III family, responsible for the generation of micro ribonucleic acids (RNAs) and the modulation of gene expression [1, 2]. Carriers are at increased risk of certain neoplasms, most occurring early in life [1, 3]. Hill et al. [4] first described a DICER1 germline pathogenic variant in familial pleuropulmonary blastoma in 2009. DICER1 syndrome, first described in 2011 [1], is also known as pleuropulmonary family tumor and dysplasia syndrome [5]. It exhibits an autosomal dominant pattern of inheritance with moderate penetrance [1], generally considered as ≤15% [3]. As many as 80% of male and 50% of female carriers may not manifest any phenotype [3], with 80% of heterozygous DICER1 germline pathogenic variants inherited and 20% de novo [5, 6] but with no clear data relating to incidence.
The spectrum of neoplasms associated with DICER1 germline pathogenic variants involves the lungs: pleuropulmonary blastoma; kidneys: cystic nephroma; female genitourinary system: ovarian sex cord-stromal tumors and embryonal rhabdomyosarcoma of the ovary/bladder/cervix; thyroid: nodular hyperplasia and differentiated thyroid cancer, and brain: pineoblastoma and pituitary blastoma [3]. Very rare linked tumors include anaplastic sarcoma of the kidney, ciliary body medulloepithelioma (dictyoma), medulloblastoma and nasal chondromesenchymal hamartoma, with T-cell Hodgkin lymphoma recently added [1, 3, 7,8,9,10,11]. Although considered, a link to Wilms tumor has not been proven [1, 12], with insufficient evidence for suggested developmental malformations pulmonary sequestration or transposition of great arteries [12]. Clinical suspicion for DICER1 syndrome typically arises in patients <6 years with pleuropulmonary blastoma or pineoblastoma and a family history of related conditions, e.g., multinodular goiter or ovarian tumors [7]. At our institution, DICER1 gene testing is recommended, and if positive, the child is recruited into a tumor surveillance program with genetic counseling, including for family members.
Pleuropulmonary blastoma is the most common DICER1 syndrome-associated tumor [1, 3]. In the International Pleuropulmonary Blastoma Registry [13], a review of 350 tumors revealed 66% had DICER1 heterozygous germline pathogenic variants. The four types of pleuropulmonary blastoma are: I and Ir (regressed): cystic, requiring only surgery with a 91% 5-year survival rate; II: cystic and solid, and III: more solid. Despite surgery and chemotherapy, pleuropulmonary blastoma types II and III have a poorer prognosis with 5-year survival rates of 71% and 53%, respectively [7, 14]. Variable distribution of DICER1 protein within pleuropulmonary blastoma may predispose to sarcomatous transformation [4]. Early diagnosis of pleuropulmonary blastoma type I is desirable to potentially avoid progression and fatal outcome [15].
Families with DICER1 germline pathogenic variants have a 10% incidence of cystic nephroma [9], a benign renal tumor usually presenting in patients <4 years old. Thin internal septae made from spindle cells are very similar to pleuropulmonary blastoma type I, raising the possibility of common pathogenesis [16]. Of 44 pediatric cystic nephromas in one review (15 with DICER1 mutations), more than 50% had a fibrous pseudocapsule, encroaching on the renal pelvis, the remainder poorly demarcated with increased internal architectural complexity [17]. Another review reported 70% of cystic nephroma patients were carriers of germline DICER1 variant carriers, with increased incidence of pleuropulmonary blastoma in childhood [18].
The International Ovarian and Testicular Stromal Tumor Registry reports a 48% incidence of DICER1 germline pathogenic variants in Sertoli-Leydig cell tumors [19]; other series report up to 60% [20]. Association with other ovarian sex cord-stromal tumor includes juvenile granulosa cell tumors and gynandroblastomas but no testicular sex cord-stromal tumors [21]. Embryonal rhabdomyosarcoma has been described as arising from the cervix, bladder and ovary [3].
Differentiated thyroid carcinoma has been described as the fourth DICER1-related malignancy [5, 22]. While this may be secondary to chemotherapy, radiotherapy or hematopoietic stem cell transplant used in the treatment of pleuropulmonary blastomas [23], Rutter et al. [24] noted an association of multinodular goiter and differentiated thyroid carcinoma in DICER1 syndrome patients even without prior chemotherapy.
Pineoblastoma is a rare neuroectodermal tumor originating in the pineal gland, and de Kock et al. [25] have demonstrated 10/21 pineoblastomas were associated with DICER1 germline pathogenic variants [3].
The purpose of this retrospective study was to review the spectrum of abnormalities manifested in DICER1 syndrome patients in a tertiary pediatric center, their imaging studies and lesion characteristics given the variable tumor phenotype. With moderate penetrance and controversy surrounding surveillance [1, 3], an additional aim was to capture modalities utilized for diagnosis, staging and surveillance and corresponding findings to better inform the guidelines for screening infants and children with DICER1 syndrome.
Materials and methods
Institutional Research Ethics Board approval was obtained for this retrospective study. This was performed at a tertiary pediatric institution in patients ≤18 years of age who underwent imaging for tumors associated with genetically confirmed or clinically suspected DICER1 syndromex between January 2004 and July 2016, including imaging prior to DICER1 germline pathogenic variant test availability in Canada (2011).
An imaging database search (ISYS Search Software Pty Ltd.; Lexmark International, Lexington, KY) using keywords pleuropulmonary blastoma, cystic nephroma, pineoblastoma, embryonal rhabdomyosarcoma, ovarian sex cord-stromal tumor, ovarian Sertoli-Leydig cell tumor and DICER1 syndrome identified the initial cohort. This search was undertaken by a pediatric radiology fellow (M.T.B.), and cross-referenced against the institutional Cancer Genetics Program and deoxyribonucleic acid (DNA) Resource Centre databases. Final diagnosis of DICER1 syndrome was obtained from the electronic patient chart based on positive genetic testing for a DICER1 germline pathogenic variant, including variants of uncertain significance with in silico evidence for pathogenicity in patients considered clinically as DICER1 syndrome and placed under surveillance based on the patients’ medical and family histories [26].
Exclusions were (1) patients with positive genetic testing but no imaging at our institution, (2) imaging acquired external to our institution during the study period and (3) patients testing negative for a DICER1 germline pathogenic variant, or in whom testing was not performed and/or the result was unknown.
Images and reports were analyzed by a pediatric radiologist (M.-L.C.G., with 16 years of post-fellowship experience) and one of two pediatric radiology fellows (M.T.B., C.M.-R.) using the institutional Picture Archive and Communication System (PACS; GE Centricity PACS Radiology RA1000 Workstation, Barrington, IL). Modalities were subclassified by body region (e.g., chest CT, brain or whole-body MRI). Also captured were the dates of the first and last examination to assess image number and frequency and surveillance duration. Imaging referrals were further analyzed, and by consensus of two reviewers (R.A.A., 2nd fellowship year, and M.-L.C.G.) were broadly categorized by indication into three groups. These were (1) primary tumor imaging for diagnosis when presenting clinically, staging, treatment/treatment complication or known tumor follow-up, (2) DICER1 surveillance imaging and (3) indications unrelated to the first two categories. Final tumor diagnosis was based on histopathology reports and/or from imaging reports and clinical follow-up. The first modality to demonstrate a lesion was assigned based on imaging reports or patient history on the electronic patient chart if relating to an external study.
Imaging was evaluated to determine the spectrum of abnormalities, anatomical location and imaging characteristics. Imaging morphology with lesion characterization was determined by two of three reviewers in consensus (M.T.B. and M.-L.C.G. or C.M.-R. and M.C.G.), the internal architecture classified as types I to III: cyst (I), septated cyst (Is), mixed cystic and solid (II) or solid (III), a macroscopic scoring system developed by the investigators (Fig. 1). Statistical analysis utilized descriptive statistics.

Graphic of imaging-based morphological classification of internal architecture of DICER1-associated neoplasms (prepared by M.T.B)
Results
Patient cohort
The imaging database search and Cancer Genetics Program and DNA Resource Centre data searches yielded 44 patients, 17 confirmed with DICER1 germline variants (11 were pathogenic and 6 were variants of uncertain significance). One patient was excluded because of a lack of available imaging. There were 12 girls and 4 boys in the final cohort of 16 with DICER1 syndrome and a mean age of 4.2 years (range: 14 days to 17 years) at first diagnostic imaging study (Table 1). DICER 1 gene testing results were negative in 7 patients, and unknown in 20. Seventy percent (14/20) of patients in the “unknown” group presented before 2011, compared with 52% in the “known” group (DICER1 negative or positive).
Radiology data
In all, 663 imaging studies were performed: 149 radiographs, 198 US, 100 CT, 161 MRI and 55 radioisotope scans. Mean duration of follow-up from the first to the last examination, or until the end of the study period, was 51.5 months (range: 4 months to 37 months) (Table 1).
Classification of imaging into one of the three groups proved to be somewhat arbitrary, with the indication not always clearly defined on the referral. On occasion, indications were overlapping such as brain MRI staging of cerebral metastases of a known primary versus brain MRI as surveillance for a pineoblastoma in DICER1 syndrome. Only if imaging was not part of primary tumor monitoring was it recorded as surveillance. With this caveat, the majority of the studies considered to be primarily DICER1 surveillance were US and MRI (Fig. 2).

Subset analysis of indication for imaging in DICER1 cohort. US ultrasound, CT computed tomography, MRI magnetic resonance imaging, NM nuclear medicine, DX imaging related to tumor diagnosis and/or staging, RX treatment-related imaging, FU monitoring of known primary malignancy
US was utilized most frequently (n=198) in 100% of patients. This consisted of combined abdominal and pelvic scans, occasionally focused renal US, with neck/thyroid scans performed in all but three patients. The majority was requested for diagnosis and/or surveillance or assessing treatment complications such as impaired renal function.
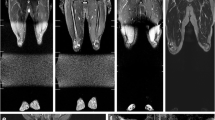
MRI was the next most frequent investigation (n=161), performed in all but one patient. While brain MRI (n=82) was the most frequent, 30 whole-body scans were performed employing a coronal single short tau inversion recovery (STIR) multistation sequence, usually but not always in combination with brain MRI (Figs. 3, 4, and 5). Whole-body MRI was performed in 62.5% (10/16) of patients (range: 1–18 per patient, commencing in 2004 with patient 5). In 80% of patients, whole-body MRI commenced from 2013, not always immediately at primary tumor diagnosis.

Example of findings on whole-body MRI using coronal short tau inversion recovery (STIR) sequence, performed as part of the surveillance protocol for patients with DICER1 syndrome, in a 23-month-old girl (patient 10) with developmental delay and a confirmed DICER1 germline pathogenic variant. The cystic and solid mass in the left lung is a pleuropulmonary blastoma first detected on this surveillance whole-body MRI (arrow)

A left cystic nephroma (arrow) is first detected on this surveillance whole-body MRI using coronal short tau inversion recovery (STIR) sequence for surveillance in a 12-year-old girl (patient 6; the third tumor shown here detected 6 years after initial presentation) with a prior history of an ovarian embryonal rhabdomyosarcoma. Renal US scan and whole-body MRI 7 months earlier were normal

Whole-body coronal short tau inversion recovery (STIR) MRI for surveillance of DICER1 syndrome in a 6-year-old boy (patient 9; this imaging 3 years after initial presentation) with previous resection of a pineoblastoma (cranial arrow) showed multiple bilateral septated renal cysts (abdominal arrows)
Plain radiographs were performed in all but one patient (n=149) and as expected, chest radiographs were most common, with a small number of abdominal and extremity radiographs and a single pre-MRI orbits radiograph. These were indicated for a combination of diagnoses, staging and/or surveillance, and management-related indications such as monitoring of central venous lines.
CT was performed less often (n=100), in 13/16 patients (81.3%). Of these, 64% of the scans incorporated chest imaging, 54.7% chest CT only, and the remainder performed in combination with neck and/or abdomen and pelvis CT.
Radioisotope scans were performed in only 43.8% of patients (n=55), usually to evaluate renal function as a consequence of primary disease or treatment complication (e.g., Tc99m-DTPA [diethylenetriaminepentacetate] scans). Rarely, other nuclear medicine studies were performed, such as [F-18]2-fluoro-2-deoxyglucose positron emission tomography, 67Gallium scans or Tc99m bone scans.
Modality choice, the timing and the frequency of imaging varied among patients and lesion types throughout the study period, including among patients with the same primary lesions.
Imaging findings
Across modalities, DICER1-related lesions were demonstrated as cystic or cystic and solid, independent of their location (Table 1). Two patients with pleuropulmonary blastomas (patients 4 and 11) presented with dyspnea and a “white-out” of one hemithorax on chest radiographs, with US , MRI and/or CT revealing pleural effusions with underlying cystic and solid masses(Fig. 6). Two patients with pleuropulmonary blastomas (patients 8 and 14) were detected only on surveillance CT, as thin-walled unilocular air-filled cysts; one pleuropulmonary blastoma (patient 10) was initially detected on whole-body MRI as a complex cystic and solid mass (Fig. 3).

Example of the imaging feature newly defined as the cracked windshield sign demonstrated across modalities is seen on a chest MRI. An axial T1 fat-saturated postcontrast sequence in a 3-year-old girl (patient 4) with a left lung mixed solid and cystic pleuropulmonary blastoma, histology confirmed on resection, demonstrates the imaging morphology classified as II – a mixed cystic and solid lesion
Pineoblastomas initially identified on CT then confirmed on MRI were large (>2 cm), lobular, pineal-based, cystic and solid enhancing lesions, often presenting with hydrocephalus – the symptoms of which prompted investigation. The one pineal cyst followed on MRI over 3 years was seen as a thin-walled unilocular cyst of <1 cm, of low signal on T1- and high signal on T2-weighted sequences, with no growth.
Cystic nephromas were larger if presenting clinically (patient 2’s measured 12.6 cm on CT and encroached on the renal pelvis [Fig. 7]), compared with surveillance imaging, with a smaller cystic nephroma in patient 6 first detected on surveillance whole-body MRI (Fig. 4). One patient (patient 5) initially shown to have renal cysts on US developed a high-grade renal sarcoma, imaging previously published, showing the cyst’s evolution from simple to septated to mixed cystic and solid morphology [9, 10]. All cystic nephromas demonstrated networks of internal septations of varying thickness on US, CT or MRI, most with pseudocapsules. In contrast, the renal sarcoma was decidedly more complex, with more solid than cystic elements on initial US.

Axial abdominal contrast-enhanced CT image of a large right septated cystic renal mass (cracked windshield sign) in a 14-month-old boy (patient 2) was pathologically confirmed to be cystic nephroma, with imaging morphology classified as Is – a septated cyst
Ovarian tumors (e.g., embryonal rhabdomyosarcoma and Sertoli-Leydig cell tumor) were first identified on regional ultrasounds as complex multiseptated cysts (Fig. 8). Affected thyroid glands were typically bulky with US demonstrating scattered anechoic cysts intermixed with echogenic nodules.

The cracked windshield imaging sign is demonstrated in a pelvic mass seen on (a) pelvic MRI (postcontrast axial fat-suppressed T1-weighted image) and (b) transverse pelvic US image in a 6-year-old girl (patient 15). This ovarian mixed solid and cystic mass (arrows) was pathologically confirmed to be a Sertoli-Leydig cell tumor, with the imaging morphology classified as II – a mixed cystic and solid lesion
Lesions such as a nasal hamartoma, seen as an homogenous opacification of the nasal cavity on CT, and fibroadenomas, first identified on whole-body MRI and confirmed with US as circumscribed oval hypoechoic breast masses, demonstrated typical appearances. Classification of all lesions, including those not associated with DICER1 syndrome, based on imaging morphology illustrated in Fig. 1 is included in Table 1.
Pathology findings
Twenty-two lesions were identified, 11 malignant tumors in 11 (68.8%) patients: 5 pleuropulmonary blastomas, 3 pineoblastomas, 1 Sertoli-Leydig cell tumor, 1 ovarian embryonal rhabdomyosarcoma and 1 high-grade renal sarcoma. No patient had more than one malignancy during the 12-year study period. Benign lesions (n=11) were present in 6 (37.5%) patients, some with multiple lesions, including thyroid cysts and nodules, renal and pineal cysts, cystic nephromas, fibroadenomas, exostoses and a nasal hamartoma. Some patients had malignant and benign lesions. Not all lesions identified have been associated with DICER1 syndrome; some such as exostoses are likely treatment-related following pelvic radiotherapy (Table 1).
Of four patients with no identified lesions, one had global developmental delay with a DICER1 germline pathogenic variant detected as part of genetic screening prompting imaging surveillance, and three were siblings of index cases or probands. Surveillance imaging commenced earlier in this group with a mean age of 0.67 years (range: 2 weeks to 22 months), compared with a mean age at tumor diagnosis and commencement of imaging of 5.09 years (range: 14 months to 17 years) (Table 1).
Discussion
To the authors’ knowledge, this is the largest imaging series in children with confirmed DICER1 syndrome, a relatively recently described entity.
Frequency of imaging
The propensity for children to develop neoplasms harboring a DICER1 germline pathogenic variant can lead to a large number of diagnostic imaging studies being performed. In our study, indications included (1) surveillance imaging from 2012; (2) oncologic diagnosis, staging and follow-up; and (3) imaging to guide therapeutic management (e.g., screening for infection, line insertions). Patient 4 had 140 investigations over 11.5 years, of which 37 were CT (23 chest CT, primarily for follow-up after pleuropulmonary blastoma was diagnosed at 3 years old) (Table 1). Our numbers may be an underestimate as one limitation of our study was that imaging performed outside our institution was not captured.
Although DICER1 syndrome does not exhibit the added sensitivity to ionizing radiation of cancer predisposition syndromes such as Li-Fraumeni syndrome [27], increased imaging frequency and the young age at which screening commences (e.g., patient 7 at 14 days) (Table 1) places them at a higher risk for radiation-related adverse effects [28, 29]. The thyroid gland is of particular concern, not infrequently in the field of view for chest CT or radiographs, and an at-risk organ in DICER1 syndrome [5, 22]. However, reflecting increased awareness in pediatric imaging to minimize ionizing radiation through use of nonionizing radiation imaging techniques and low dose techniques in CT (beyond the scope of this study), US then MRI were performed most frequently (Table 1).
Spectrum of pathology
Our understanding of the variable phenotype of DICER1 syndrome continues to unfold. The range of neoplasms in our patient cohort was as expected, with pleuropulmonary blastomas and pineoblastomas being most frequent [3, 14,15,16,17,18,19,20,21,22]. However, the prevalence of malignant lesions in our patient cohort (68.8%) was higher than anticipated (Table 1). Similarly, the organs involved were also as previously described [2, 13,14,15,16,17,18,19,20,21]. This emphasizes that this combination of tumors and organs is an important red flag in considering a diagnosis of DICER1 syndrome (Fig. 9).

Target organs involved in DICER1 syndrome: (1) thyroid gland, (2) kidney, (3) pineal gland, (4) lung and (5) ovary (prepared by R.A.A.)
The higher prevalence of malignant neoplasms likely reflects our methodology, using an imaging database search for patient identification thus introducing a selection bias of patients with lesions already under investigation. However, this was countered by cross-referencing with the Cancer Genetics Program and DNA Resource Centre databases, identifying six additional patients, with available imaging for five of them. Referral bias of a tertiary care centre may also have influenced this high prevalence.
Conversely, we may be underestimating the DICER1 syndrome cohort at our institution. Twenty of 44 patients identified from the imaging database search with DICER1 syndrome-associated lesions were excluded because their DICER1 germline pathogenic variant status was unknown. However, not all unknowns are likely to have DICER1 syndrome as 7 of 44 identified from the imaging database search were excluded because of negative DICER1 gene testing. Multinodular goiter was not a specific keyword imaging database search as it was considered likely to have a low yield for DICER1 syndrome given the number of thyroid lesions in the general pediatric population. Regardless, thyroid nodules and cysts were identified in the patient cohort, some of which were biopsied (Table 1).
Only three DICER1 positive relatives of probands were included, none with tumors in the study period. This, along with the small sample size, constitutes further limitations with respect to assessing tumor prevalence.
Imaging characteristics
The majority of cystic/septated lesions were benign and those with solid components were malignant. A similar imaging pattern was observed among tumors affecting different organs suggesting a common pathological origin, with one example of cystic lesions progressing into more solid tumors, with a correspondingly worse prognosis. Doros et al. [30] have recently described a similar observation from a pathological standpoint in a cohort of 20 cystic nephromas and 6 cystic partially differentiated nephroblastomas, with 4 cases of high-grade renal sarcomas with residual cystic nephroma adjacent to the malignant component. They hypothesized a common model of progression from cysts to high-grade sarcomas. Of note, no Wilms tumors were identified in our cohort.
Early diagnosis of these tumors at a simpler cystic stage is paramount due to the increasingly unfavorable prognosis of pleuropulmonary blastomas types II and III or anaplastic renal sarcomas, thus highlighting the important role of effective surveillance strategies in reducing morbidity and mortality [14]. Given the early onset of pleuropulmonary blastomas and cystic nephromas, it becomes extremely important to differentiate these tumors from other benign cystic conditions of infancy. Preoperatively distinguishing cystic congenital pulmonary airway malformation from predominantly cystic pleuropulmonary blastomas in the lungs can be challenging, warranting further research [31]. As illustrated by Kousari et al. [32], pleuropulmonary blastoma or cystic nephroma in a patient with a confirmed diagnosis of DICER1 syndrome should prompt imaging for concurrent lesions. The paucity of literature addressing management of pineal cysts or ovarian follicles in patients with DICER1 syndrome is an added complication.
In our study, we observed a common appearance to these tumors – some with thin internal septations within a cyst, others with a central hub and multiple disorganized radial threads, resembling a cracked windshield. This pattern was demonstrated in lesions occurring in different organs across modalities: US, CT and MRI (Figs. 6, 7, and 8). As such, we propose a new radiologic sign, the cracked windshield sign, denoting this pattern of cystic lesion in relevant organs, alerting us to the possibility of DICER1 syndrome in children. Recognizing this sign may be a useful tool in earlier diagnosis of at-risk lesions, although it requires validation in a larger patient cohort to determine if it is specific to DICER1-related tumors.
Surveillance imaging in DICER1 syndrome
Surveillance in DICER1 syndrome remains controversial. In detailing the modest penetrance and variable phenotype of DICER1 syndrome in 2011, Slade et al. [1] alluded to the complexity this created for surveillance, opting for an open-door management policy: early investigation of tumor-related symptoms, no routine surveillance in healthy mutation-positive individuals. In 2016, the complexity remains. However, our findings suggest surveillance, in the appropriate clinical context (i.e. one DICER1-associated neoplasm particularly of early onset or >1 in the patient and/or close relative) to prompt genetic testing confirming DICER1 pathogenic variants, is of value in children for detecting clinically occult tumors and/or monitoring lesion evolution.
Special care should be given to the frequency and mode of imaging in infants and children to minimize potential new risks such as ionizing radiation by preferentially using nonionizing techniques, as in our study with US and MRI more commonly performed than radiographs, CT or radioisotope scans. Screening introduces other costs -- financial and psychological -- to be weighed in the balance given their impact on patients and their families.
The institutional surveillance protocol was introduced in 2012, evolved over time and was variably implemented by our clinicians. As in any surveillance protocol, particularly in newly recognized disease processes such as DICER1 syndrome, regular review was undertaken to understand the risks and benefits [33]. Our local practice changed in response to our experience with DICER1 syndrome and in response to available published data.
During the study period, the range of modalities utilized was relatively constant but differed widely among patients (Table 1). Chest surveillance (chest radiographs 6 monthly to age 8 years, 12 monthly to age 18 years, plus initial CT chest +/− follow-up) was variably implemented. This also varied with tumor diagnosis. Wide differences in the use of US were also observed. Initial surveillance with 6-monthly renal US (for cystic nephroma) expanded to include pelvic (for ovarian Sertoli-Leydig cell tumors and embryonal rhabdomyosarcoma) and thyroid US (for nodular thyroid hyperplasia/carcinoma), at 6 and 12 months respectively to age 8 years, although not implemented in all patients. Brain MRI (for pineoblastoma) was performed in 87.5% of patients, not all annually.
While whole-body MRI was not part of the original surveillance protocol, 60% of patients had whole-body MRI, mostly after 2013. Three (13.6%) lesions were initially identified on whole-body MRI – pleuropulmonary blastoma, cystic nephroma (Figs. 3 and 4) and breast fibroadenoma. There is increasing evidence of the utility of whole-body MRI in screening pediatric cancer predisposition syndrome patients [34], with advantages including the lack of ionizing radiation and a single modality one-stop shop. Interweaving annual whole-body MRI, with chest radiographs and the already specified regional US imaging, at 12-month intervals, possibly replacing chest CT if initial CT is negative, warrants consideration. However, this must be weighed against any motion artifact in younger patients necessitating general anesthesia, at our site typically <6 years, a valid consideration given the mean age at first imaging in our cohort was 4.2 years.
DICER1 germline testing only became available in Canada in 2011, with DICER1 syndrome recognized around that time, and likely led to greater awareness of the spectrum of DICER1 syndrome-associated neoplasms with progressive, although variable, uptake of the surveillance protocol.
Moving forward, it is anticipated that our institutional surveillance protocol may be further modified to incorporate guidelines forthcoming from international DICER1 workshops held in May and September 2016. These were convened in part to establish recommendations for testing and surveillance guidelines for individuals with DICER1 pathogenic variants.
Conclusion
The spectrum and evolution of DICER1-related tumors and the young age at presentation suggest early surveillance of at-risk patients is critical, while minimizing exposure to ionizing radiation. Awareness of this constellation of tumors, involving the lungs, kidneys, pineal and thyroid glands, and ovaries, and their potential to evolve, warrants close observation. This is particularly so for lesions manifesting the cracked windshield sign. Pediatric radiologists have a central role to play in identifying at-risk patients, particularly when lesions coexist, coordinating integrated imaging for diagnosis and surveillance.
References
Slade I, Bacchelli C, Davies H et al (2011) DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 48:273–278
Online Mendelian Inheritance in Man. National Center for Biotechnology Information website. http://www.ncbi.nlm.-nih.gov/omim. (http://www.omim.org/entry/606241). Accessed 27 May 2016
Foulkes WD, Priest JR, Duchaine TF (2014) DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14:662–672
Hill DA, Ivanovich J, Priest JR et al (2009) DICER1 mutations in familial pleuropulmonary blastoma. Science 325:965
Online Mendelian Inheritance in Man. National Center for Biotechnology Information website. http://www.ncbi.nlm.-nih.gov/omim. (http://www.omim.org/entry/601200). Accessed 27 May 2016
Priest JR, Watterson J, Strong L et al (1996) Pleuropulmonary blastoma: a marker for familial disease. J Pediatr 128:220–224
Doros L, Schultz KA, Stewart DR et al (2014) DICER1-related disorders. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews® [Internet]. University of Washington, Seattle, Seattle (WA), pp 1993–2016 Available at: https://www.ncbi.nlm.nih.gov/books/NBK196157/. Accessed 27 May 2016
de Kock L, Druker H, Weber E et al (2015) Ovarian embryonal rhabdomyosarcoma is a rare manifestation of the DICER1 syndrome. Hum Pathol 46:917–922
Wu MK, Goudie C, Druker H et al (2016) Evolution of renal cysts to anaplastic sarcoma of kidney in a child with DICER1 syndrome. Pediatr Blood Cancer 63:1272–1275
Wu MK, Cotter MB, Pears J (2016) Tumor progression in DICER1-mutated cystic nephroma - witnessing the genesis of anaplastic sarcoma of the kidney. Hum Pathol 53:114–120
Kuhlen M, Hönscheid A, Schemme J et al (2016) Hodgkin lymphoma as a novel presentation of familial DICER1 syndrome. Eur J Pediatr 175:593–597
Foulkes WD, Bahubeshi A, Hamel N et al (2011) Extending the phenotypes associated with DICER1 mutations. Hum Mutat 32:1381–1384
The International Pleuropulmonary Blastoma Registry. http://www.ppbregistry.org. Accessed 27 May 2016
Messinger YH, Stewart DR, Priest JR et al (2015) Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the international Pleuropulmonary Blastoma registry. Cancer 121:276–285
Schultz KA, Harris A, Williams GM et al (2014) Judicious DICER1 testing and surveillance imaging facilitates early diagnosis and cure of pleuropulmonary blastoma. Pediatr Blood Cancer 61:1695–1697
Bahubeshi A, Bal N, Rio Frio T et al (2010) Germline DICER1 mutations and familial cystic nephroma. J Med Genet 47:863–866
Cajaiba MM, Khanna G, Smith EA et al (2016) Pediatric cystic nephromas: distinctive features and frequent DICER1 mutations. Hum Pathol 48:81–87
Faure A, Atkinson J, Bouty A et al (2016) DICER1 pleuropulmonary blastoma familial tumour predisposition syndrome: what the paediatric urologist needs to know. J Pediatr Urol 12:5–10
Schultz KA, Harris A, Messinger Y et al (2016) Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Familial Cancer 15:105–110
Heravi-Moussavi A, Anglesio MS, Cheng SW et al (2012) Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 366:234–242
Conlon N, Schultheis AM, Piscuoglio S et al (2015) A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol 28:1603–1612
Oue T, Inoue M, Kubota A et al (2008) Pediatric thyroid cancer arising after treatment for pleuropulmonary blastoma. Pediatr Blood Cancer 50:901–902
de Kock L, Sabbaghian N, Soglio DB (2014) Exploring the association between DICER1 mutations and differentiated thyroid carcinoma. J Clin Endocrinol Metab 99:E1072–E1077
Rutter MM, Jha P, Schultz KA et al (2016) DICER1 mutations and differentiated thyroid carcinoma: evidence of a direct association. J Clin Endocrinol Metab 101:1–5
de Kock L, Sabbaghian N, Druker H et al (2014) Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 128:583–595
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Kleinerman RA (2009) Radiation-sensitive genetically susceptible pediatric sub-populations. Pediatr Radiol 39(Suppl 1):S27–S31
Brenner DJ, Shuryak I, Einstein AJ (2011) Impact of reduced patient life expectancy on potential cancer risks from radiologic imaging. Radiology 261:193–198
Brady Z, Ramanauskas F, Cain TM et al (2012) Assessment of paediatric CT dose indicators for the purpose of optimisation. Br J Radiol 85:1488–1498
Doros LA, Rossi CT, Yang J et al (2014) DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 27:1267–1280
Oliveira C, Himidan S, Pastor AC et al (2011) Discriminating preoperative features of pleuropulmonary blastomas (PPB) from congenital cystic adenomatoid malformations (CCAM): a retrospective, age-matched study. Eur J Pediatr Surg 21:2–7
Kousari YM, Khanna G, Hill DA et al (2014) Case 211: pleuropulmonary blastoma in association with cystic nephroma-DICER1 syndrome. Radiology 273:622–625
Chong AL, Grant RM, Ahmed BA et al (2010) Imaging in pediatric patients: time to think again about surveillance. Pediatr Blood Cancer 55:407–413
Anupindi SA, Bedoya MA, Lindell RB et al (2015) Diagnostic performance of whole-body MRI as a tool for cancer screening in children with genetic cancer-predisposing conditions. AJR Am J Roentgenol 205:400–408
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
Bueno, M.T., Martínez-Ríos, C., la Puente Gregorio, A.D. et al. Pediatric imaging in DICER1 syndrome. Pediatr Radiol 47, 1292–1301 (2017). https://doi.org/10.1007/s00247-017-3875-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-017-3875-0