
Abstract
Background
Thanatophoric dysplasia (TD) and hypochondroplasia are both caused by FGFR3 (fibroblast growth factor receptor 3) gene mutations. Temporal lobe dysplasia has been well described in thanatophoric dysplasia; however, only a couple of anecdotal cases of temporal lobe dysplasia in hypochondroplasia have been described.
Objective
To define temporal lobe abnormalities in patients with hypochondroplasia, given that they share the same genetic mutation.
Materials and methods
We identified brain imaging studies of nine children with hypochondroplasia. The temporal lobes were assessed on CT and MRI for size and configuration of the temporal horn and aberrant sulcation of the inferior surface of the temporal lobe.
Results
All children had a triangular-shape temporal horn and deep transverse fissures of the inferior temporal lobe surface. Neuroimaging in our cohort revealed enlarged temporal lobes and oversulcation of the mesial temporal and occipital lobes, with abnormal inferomedial orientation of these redundant gyri. Hippocampal dysplasia was also universal.
Conclusion
We confirmed frequent inferomesial temporal and occipital lobe abnormalities in our cohort of children with hypochondroplasia. Murine models with mutant fgfr3 display increased neuroprogenitor proliferation, cortical thickness and surface area in the temporo-occipital cortex. This is thought to result in excessive convolution and likely explains the imaging findings in this patient cohort. (Note that fgfr3 is the same genetic mutation in mice as FGFR3 is in humans.)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypochondroplasia, achondroplasia and thanatophoric dysplasia (TD) are skeletal conditions of rhizomelic short stature that have in common mutations of the fibroblast growth factor receptor 3 gene (FGFR3). The phenotypic spectrum reflects the severity of the underlying genetic mutations, with hypochondroplasia being the mildest phenotype and TD the lethal form. Achondroplasia is characterized by rhizomelic short stature, macrocephaly, trident hands and characteristic facial features that include frontal bossing. Features are consistent, which reflects the relative genetic homogeneity of the disorder [1] (a Glyc380Arg substitution in the FGFR3 sequence). Hypochondroplasia manifests similar though milder clinical and radiologic features than achondroplasia and molecular linkage studies now support allelism of these conditions [1]. The characteristic clinical features are short stature, rhizomelic arms and legs, lumbar lordosis and macrocephaly [1, 2]. Radiologic features include shortening of long bones with mild metaphyseal flare, narrowing of inferior lumbar interpedicular distances, shortening of lumbar pedicles and distal ulna, short broad femoral neck and squared, short ilia [2]. Two gene mutations, 1620C>A and 1620C>G, which result in a lysine for arginine substitution at codon 540 (N540K), account for the majority of cases [2]. However, these mutations do not occur in all cases, which accounts for the clinical heterogeneity of this condition [1]. TD demonstrates more severe forms of features common with achondroplasia, such as shortening of the long bones and macrocephaly. However, other characteristic features such as platyspondyly, bowed femurs, cloverleaf skull and severely shortened ribs are also seen in TD, the last resulting in perinatal lethality.
Temporal lobe abnormalities associated with TD have been well documented [3–6]. Megalencephaly, temporal lobe enlargement and deep fissures orientated transversely across the inferomedial aspects of the temporal lobes are seen on imaging [5]. The features reported on neuropathology are megalencephaly, hippocampal dysplasia, rudimentary dentate gyrus and polymicrogyria [3]. Commonly identified features include temporal lobe enlargement (Fig. 1), deep transverse temporal sulci, subependymal neuronal heterotopia and subarachnoid glial heterotopia (75%). The cortical malformations are seen as early as mid-gestation [3, 6], before the development of craniosynostosis, confirming the dysplastic nature of these abnormalities rather than deformation caused by skull base anomalies. These abnormalities are manifested on neuroimaging [5]. Given the common genetic and clinical features, it is not surprising that similar mesial temporal lobe abnormalities have been reported anecdotally in hypochondroplasia [7, 8]. We identified the prevalence and features of temporal lobe abnormalities in our patient population with hypochondroplasia.

Coronal STIR MR image shows enlargement of the temporal lobe in a 1-week-old male with hypochondroplasia, as demonstrated by bulging of the surface of the temporal lobe beyond the contour of the adjacent frontoparietal lobe (arrow)
Materials and methods
Patients
After obtaining approval from the institutional ethics board, we searched our PACS database (1995–2013) using a radiology text word-search programme, ISYS™, for children with hypochondroplasia who had brain imaging performed. We reviewed the clinical notes of these children and included those with a clinical diagnosis of hypochondroplasia recorded by the senior geneticist.
Clinical data
We reviewed the medical records of all children and recorded all pertinent clinical information, particularly the presence of epilepsy and apnoeic episodes, and we made note of head circumference. We also recorded the results of genetic testing.
Imaging
We reviewed all US, CT and MR images for each child and recorded the imaging modalities, MR sequences, dates and ages of the children at the time of imaging.
Data
Two paediatric neuroradiologists reviewed the available imaging for each child and examined the configuration of the temporal and occipital lobes. The appearance of the hippocampus, dentate gyrus, amygdala (Fig. 2) and temporal and medial occipital lobes was described and recorded. We also searched for other specific features including the presence of deep transverse sulci, megalencephaly, temporal lobe enlargement (defined as bulging of the surface of the temporal lobe beyond the contour of the adjacent frontoparietal lobe, Fig. 1), polymicrogyria, subependymal neuronal heterotopia and subarachnoid glial heterotopia. We examined the grey–white matter differentiation in the mesial temporal lobes. We recorded the presence of ventriculomegaly, which was defined as atrial measurement >10 mm (using fetal imaging standards not formally established for postnatal imaging [9]). The images were evaluated independently with consensus reached at the workstation.

Coronal T2-W MRI shows abnormal globular shape of the amygdala in an 11-year-old male with hypochondroplasia
Results
Patients
Nine children had a clinical diagnosis of hypochondroplasia. Children ranged in age at first diagnostic imaging from 1 day to 4 years, with a median age 10.5 weeks.
Clinical data
Two children had neonatal seizures and one child had apnoeic episodes since birth. One had mild gross motor developmental delay. All children had macrocephaly, (>95th centile for age and gender) apart from one extremely premature infant (28 weeks’ gestation). One child had a ventriculoperitoneal shunt inserted at 18 months of age for hydrocephalus. Genetics testing had been performed in two children and was positive for the common FGFR3 Asn540Lys mutation. Testing was deemed unnecessary in one child in whom the father had confirmation of the common genetic mutation. The remaining five children had not had genetics testing because it was either not available at the time of their diagnosis or not indicated in the presence of a positive skeletal survey. There was one child in whom the clinical diagnosis was suspected but genetics testing for the known mutations was negative.
Imaging
Seven children had MRI studies, three had CT and MRI studies, two had US and MR studies, and two had only CT studies (Table 1).
The prevalence of features is outlined in Table 2. Megalencephaly, (diffuse enlargement of the brain) was found in all children apart from the extremely premature child. Mild ventriculomegaly, which was measured as an atrial diameter on axial plane of 10–12 mm [9], was found in 7/9 children. Imaging studies in all children demonstrated abnormal triangular temporal horn configuration (Fig. 3). Relative temporal lobe enlargement was found in all children who had MRI studies (Fig. 1), though it could not be confirmed in the two children in whom only CT was performed because coronal reformats were not available.

Temporal horn and mesial temporal lobe configurations. a Axial T1-W MRI shows the normal configuration of temporal horns and mesial temporal lobes in a 7-year-old female. b Axial T1-W MRI shows abnormal triangular configuration of temporal horns and deep transverse clefts of mesial temporal lobes in a 7-year-old female with hypochondroplasia
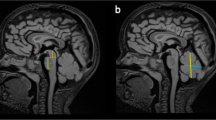
Deep transverse clefts in the mesial temporal lobe (Fig. 3) were found in all children. These clefts were orientated medially, towards the circum mesencephalic cisterns, and were apparent on sagittal slices through the mesial temporal lobes (Fig. 4). Oversulcation of the inferomedial temporal lobes, with abnormal inferomedial orientation of these redundant gyri, was a consistent finding in children in whom an MRI was performed (7/7) (Fig. 5). This appearance extended to involve the medial occipital lobes and calcar avis in all children (Fig. 6). One of our patients also demonstrated subependymal heterotopia, which has been described in TD.

Clefting of the mesial temporal lobe. a Sagittal T2-W MR image shows a normal mesial temporal lobe in a 2-year-old male. b Sagittal T2-W image shows abnormal sagittal clefting in the mesial temporal lobe in a 2-year-old male with hypochondroplasia

Sulcation of the mesial temporal lobes. a Coronal T2-W MR image shows normal sulcation and grey–white matter differentiation of the mesial temporal lobes in a 16-year-old female. b Coronal T2-W image shows oversulcation of the mesial temporal lobes and lack of grey–white matter differentiation in a 16-year-old female with hypochondroplasia

Sulcation of the calcar avis. a Axial T1-W MRI shows normal sulcation of the mesial temporo-occipital lobes and calcar avis in a 7-year-old female. b The same sequence shows oversulcation extending to the calcar avis in a 7-year-old female with hypochondroplasia
Hippocampal dysplasia and hypoplastic dentate gyrus were also identified in all children in whom an MRI was performed (7/7) (Fig. 7). Grey matter heterotopia was found in one child.

Hippocampal dysplasia and hypoplastic dentate gyrus. a Coronal T2-W MRI shows normal formation of the hippocampus and dentate gyrus in a 16-year-old. b Hippocampal dysplasia and hypoplastic dentate gyrus are seen on coronal T2-W MR in a 16-year-old with hypochondroplasia
Discussion
Abnormal temporal lobes were first described by Grosso et al. [8] in a 5-year-old with hypochondroplasia and FGFR3 Asn540Lys mutation who presented with complex partial seizures at 2 months of age. The authors noted inadequate grey–white matter differentiation, abnormal configuration of hippocampal structures and temporal horns and mild ventriculomegaly. On review of the images, we additionally identified transverse clefts of the inferior surfaces of the temporal lobes. Since then there have been reports of three patients with the same genetic mutation who have dysplastic hippocampi and parahippocampal gyri, loss of grey–white matter differentiation, abnormal temporal horn configuration and unusual temporal lobe configuration [7, 10]. We have illustrated similar imaging findings in our patient cohort and have noted additionally the presence of deep transverse clefts, abnormal temporal horns and oversulcation of the mesial temporal lobes.
All patients described in the cases published to date had apnoeic episodes in infancy, two with complex partial seizures in infancy and one with additional autism and developmental delay. We found similar clinical features in our cohort, though with reduced frequency.
Mild hydrocephalus was a common finding, with one child requiring shunting. Although hydrocephalus is also common in TD it is not a constant feature, and it is thought that mild temporal horn enlargement results from expansion of the temporal lobe rather than obstruction [3]. The enlarged ventricles may therefore reflect megalencephaly rather than obstruction.
Hippocampal dysplasia, rudimentary dentate gyrus, enlarged temporal lobes and abnormal sulcation are core neuropathological features of TD, evident at 19–20 gestational weeks [3, 5, 6]. We have illustrated similar imaging findings in hypochondroplasia, supporting evidence from animal studies where fgfr3 mutations have resulted in a specific temporo-occipital lobe phenotype.
FGFR3 gene regulates chondrocyte proliferation and differentiation and is also instrumental in cortical patterning and neurogenesis [3]. The specific molecular genetic defect is heterogeneous in type 1 TD (characterised by platyspondyly and curved femurs), although all types have mutations of FGFR3 [11]. The sequence affected in type 2 TD (characterised by the cloverleaf skull), pLys650Glu, encodes a protein product that confers stability to a tyrosine kinase cell surface receptor [11]. When this sequence is aberrant, constitutive activation of receptor tyrosine kinase and downstream signalling results in increased cell proliferation and differentiation [3, 12]. In the murine model of TD with conditional knockout in gene fgfr3+/K644E this mutation results in enlargement of the brain, increased total cell number and proportionately thicker cortical layers [13]. It has been shown that this enlargement and cortical thickness, particularly within the cortical plate and intermediate zone, predominates in the temporo-occipital cortex, correlating with the gradient of fgfr3 expression in the ventricular zone at early stages of neurogenesis, and results from increased numbers of intermediate progenitors [14]. The major limitation of this mouse model is that mice do not normally sulcate; however, it is believed that the selective surface expansion observed in mice is likely to reflect the premature gyrification of the temporo-occipital cortex in human TD. This study confirms pathology results from early regional increase in surface area [14]. In addition, there is reduced expression of a cortical hem marker, Wnt2b, observed in fgfr +/K644E primordium [14]. Because the cortical hem determines growth and patterning for the adjacent hippocampal primordium [3], this might account for preliminary observations of hippocampal dysplasia and reduction in size of the CA3/dentate gyrus in the mice models [14].
Conclusion
We have demonstrated consistent mesial temporal and occipital lobe abnormalities in a cohort of children with a diagnosis of hypochondroplasia, abnormalities that are similar to those described in the more severe phenotypic variant TD with a common association of FGFR3 gene mutation. Our findings therefore suggest the importance of the FGFR3 gene in development and neuronal organization of the temporo-occipital cortex in humans. This has important clinical implications in early diagnosis, genetic counselling and management of seizure-related disorders in these conditions.
References
Vajo Z, Francomano CA, Wilkin DJ (2000) The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, Muenke craniosynostosis, and Crouzon syndrome with acanthosis nigricans. Endocr Rev 21:23–39
Francomano CA et al (1999) Hypochondroplasia. In: Pagon RA, Bird TD, Dolan CR (eds) GeneReviews (Internet). University of Washington, Seattle, Updated 2005. http://www.ncbi.nlm.nih.gov/books/NBK1477/
Hevner RF (2005) The cerebral cortex malformation in thanatophoric dysplasia: neuropathology and pathogenesis. Acta Neuropathol 110:208–221
Coulter CL, Leech RW, Brumback RA et al (1991) Cerebral abnormalities in thanatophoric dysplasia. Childs Nerv Syst 7:21–26
Miller E, Blaser S, Shannon P et al (2009) Brain and bone abnormalities of thanatophoric dwarfism. AJR Am J Roentgenol 192:48-51
Knisely AS, Ambler MW (1988) Temporal-lobe abnormalities in thanatophoric dysplasia. Pediatr Neurosci 14:169–176
Kannu P, Hayes IM, Mandelstam S et al (2005) Medial temporal lobe dysgenesis in hypochondroplasia. Am J Med Genet A 138:389–391
Grosso S, Farnetani MA, Berardi R et al (2003) Medial temporal lobe dysgenesis in Muenke syndrome and hypochondroplasia. Am J Med Genet A 120A:88–91
Senapati GM, Levine D, Smith C et al (2010) Frequency and cause of disagreements in imaging diagnosis in children with ventriculomegaly diagnosed prenatally. Ultrasound Obstet Gynecol 36:582–595
Kannu P, Aftimos S (2007) FGFR3 mutations and medial temporal lobe dysgenesis. J Child Neurol 22:211–213
Francomano CA (1999) Hypchondroplasia. In: Pagon RA, Bird TD, Dolan CR et al (eds) GeneReviews (Internet). University of Washington, Seattle, Available via http://www.ncbi.nlm.nih.gov/books/NBK1477
Iwata T, Hevner RF (2009) Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ 51:299–323
Inglis-Broadgate SL, Thomson RE, Pellicano F et al (2005) FGFR3 regulates brain size by controlling progenitor cell proliferation and apoptosis during embryonic development. Dev Biol 279:73–85
Thomson RE, Kind PC, Graham NA et al (2009) Fgf receptor 3 activation promotes selective growth and expansion of occipitotemporal cortex. Neural Dev 4:4
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Philpott, C.M., Widjaja, E., Raybaud, C. et al. Temporal and occipital lobe features in children with hypochondroplasia/FGFR3 gene mutation. Pediatr Radiol 43, 1190–1195 (2013). https://doi.org/10.1007/s00247-013-2684-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-013-2684-3