
Abstract
Tuberous sclerosis is a complex autosomal-dominant neurocutaneous syndrome characterized by hamartomatous malformations of fibrous and connective tissues in various organs. Although various histologic types of soft-tissue masses can occur with tuberous sclerosis, we present a unique case of fibrous hamartoma of infancy presenting as large infiltrating cutaneous and subcutaneous masses in the abdominal wall in a 4-year-old boy with tuberous sclerosis. Although the co-occurrence of tuberous sclerosis and fibrous hamartoma of infancy is very rare, it should be considered in the differential diagnosis of subcutaneous soft-tissue masses found in children with tuberous sclerosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tuberous sclerosis (TS) is a rare autosomal-dominant disorder characterized by hamartomatous malformations of fibrous and connective tissue in various organs such as the brain, kidney, heart, and lung [1]. Occasionally, the characteristic cutaneous features of TS are helpful in the early detection of visceral involvement. Facial angiofibroma, ungual fibroma and shagreen patch are the most common cutaneous lesions [2].
Various histologic types of soft-tissue masses or hamartomas involving solid organs can occur in patients with TS. However, fibrous hamartoma of infancy presenting as a large infiltrating cutaneous and subcutaneous abdominal wall mass in a patient with TS is a unique finding.
We describe a case of fibrous hamartoma of infancy involving the abdominal wall in a patient with known TS. Although fibrous hamartoma has a completely benign course, because of its rapid infiltrative growth it is very important to properly recognize this tumor as soon as possible in order to excise it completely [3].
Case report
A 4-year-old boy presented with an 18-month history of a soft-tissue mass on the anterolateral side of the left abdominal wall. The lesion had rapidly grown to 12×6.5 cm at the time of presentation. This rapid growth had alarmed the child’s parents. The boy had been diagnosed with and treated for TS from birth based on the clinical features, including epilepsy and mental retardation, and multiple subependymal nodules and cortical tubers seen on brain MRI (not shown).
Physical examination revealed a firm nonmobile mass that appeared to originate from subcutaneous tissue in the left abdominal wall (Fig. 1). The mass had a smooth surface without any inflammatory signs such as tenderness, erythema or warmth.

The gross findings of fibrous hamartoma of infancy. a The skin surface has a nodular appearance because of the underlying lesion composed of skin and the subcutaneous layers, which, on gross examination, appeared yellowish and multinodular. b The cut section shows ill-defined whitish-gray fibrotic strands admixed with yellow fat in the deep dermis. The well-defined oval mass is the epidermal inclusion cyst
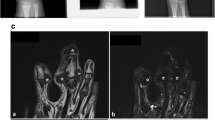
CT scan revealed an ill-defined infiltrative mass involving the anterolateral wall of the left abdomen; it was mainly situated in subcutaneous tissue and muscle layers. The mass was composed of mixed soft-tissue and fat densities without peritoneal invasion. Other multiple well-defined oval-shaped hypodense nodules were scattered in the skin layer of the left anterolateral abdominal wall (Fig. 2). MR scan also identified a mixed inhomogeneous pattern consisting of fat and intermediate-intensity tissues that were hypointense and intermediate signal intensities on T1-W and T2-W imaging. There were spotty high signal intensities with fat suppression on both sequences. Multiple well-defined oval-shaped nodules were distributed in the skin layer. These nodules showed high signal intensities on the T2-W sequence and intermediate signal intensities on the T1-W sequence, and there was no evidence of enhancement (Fig. 3).

CT image obtained after intravenous administration of contrast agent reveals a bulky infiltrative mass involving the left anterolateral abdominal wall. The mass is distributed primarily throughout the subcutaneous and muscle layers, and is composed of multiple nodular, soft-tissue and fat densities. There is no peritoneal invasion. Other well-defined oval-shaped hypodense nodules involve the skin layer (arrows)

MR images. Axial T1-W (a), T2-W (b) and fat-suppressed enhanced T1-W (c) images of the abdomen show an inhomogeneous pattern consisting of fat and intermediate intensity tissue. There are spotty high signal intensities on the T1-W image and the T2-W image with fat suppression on the fat-suppressed enhanced T1-W image, and images are isointense on T1-W and mildly hypointense to muscle on T2-W imaging with mild enhancement (straight arrows). There are serpentine low-signal intensities on the T1-W and T2-W images (arrowheads). Multiple well-defined oval-shaped nodules are distributed in the skin layer. These nodules show high signal intensity on the T2-W image and intermediate signal intensity on the T1-W sequence, and there is no evidence of enhancement (curved arrows)
Based on the imaging features, we thought the tumor might be one of the fibromatoses seen in children, or plexiform neurofibromatosis. The mass was excised. The resected specimen, which was composed of skin and subcutaneous tissue, appeared yellowish and multinodular (Fig. 1). On a cut section, ill-defined whitish-gray fibrotic strands were admixed with yellow fat in the deep dermis. Multiple well-defined oval nodules were diagnosed as epidermal inclusion cysts (Fig. 1).
On histopathologic examination, the soft-tissue mass was seen to be composed of well-formed fibrous trabeculae, islands of immature mesenchymal cells, and mature adipose tissue (Fig. 4).High-power view of this specimen showed trabecules composed of dense fibrocollagenous tissue (Fig. 4). Mature adipose tissue was admixed between the fibrous trabeculae (Fig. 4); however, neither nuclear atypia nor mitotic figures were found. Histologic examination confirmed the diagnosis of fibrous hamartoma of infancy.

Histopathologic examination. a The lesion demonstrates well-formed fibrous trabeculae, islands of immature mesenchymal cells, and mature adipose tissue (H&E, ×100). b High-power view (H&E, ×400) shows trabeculae composed of dense fibrocollagenous tissue (arrows). Mature adipose tissue (asterisks) is admixed between fibrous trabeculae. Histologic examination confirmed the diagnosis of fibrous hamartoma of infancy
Discussion
TS is a rare autosomal-dominant neurocutaneous syndrome characterized by hamartomatous malformations of fibrous and connective tissues in various organs [1]. The involvement of vital organs such as the brain, kidney, heart, and lung is the main cause of death in patients with TS [2]. In TS patients, inactivation of the tuberous sclerosis complex tumor-suppressor genes TSC1 and TSC2 contributes to the development of a wide range of hamartomatous lesions [4]. The genetic tendency for TS to give rise to hamartomas might explain the coexistence of TS and hamartoma. However, fibrous hamartoma of infancy has no known familial or syndromic associations [5]. More detailed studies are needed to prove a genetic link between the fibrous hamartoma of infancy and TS.
Fibrous hamartoma of infancy is a benign and myofibroblastic proliferative subcutaneous tumor [6]. It is usually diagnosed within the first 2 years of life (at a median age of 10 months), and in nearly 25% of patients it is congenital. It is found predominantly in boys [6]. The common sites of involvement are the axillary regions, shoulder, upper arms, and trunk; however, lesions have been found in the extremities, inguinal region, and the external genital areas [6].
Frequently, fibrous hamartomas present as solitary subcutaneous masses that are poorly demarcated from the adjacent soft tissue and are contiguous with the surrounding fat [5]. Adjacent structures can be infiltrated, and local resection of the lesion is usually curative, with a low risk of recurrence. MRI findings for fibrous hamartoma of infancy are characteristic, i.e. strands of intermediate signal intensity interspersed with fat [6, 7].
In our patient primarily low to intermediate signal intensities were seen on both T1- and T2-W MRI sequences. These findings, which are rare in a soft-tissue tumor, indicated that the tumor had a rich fibrous component. Spotty high intensities on both sequences and the fat suppression on the fat-suppressed T1-W image corresponded to the abundant fat content. These imaging findings are common for other pediatric soft-tissue tumors composed of adipose tissue and fibroblastic elements [6, 8]. The differential diagnoses in these cases include fibrous tumors of infancy and childhood, and plexiform neurofibromatosis. In addition, congenital fibrosarcoma can have a similar MRI appearance to that of infantile fibromatosis. Thus histologic confirmation is needed to establish the differential diagnosis [6].
The fibromatoses are a group of disease processes characterized by benign proliferation of fibrous tissue with histology and behavior mimicking malignancy [8]. Among these fibromatoses, fibrous hamartoma of infancy, infantile fibromatosis, myofibromatosis and congenital fibrosarcoma should be included in the differential diagnoses. Infantile fibromatoses are usually poorly circumscribed and show a high risk of recurrence. MRI shows intermediate to low signal intensity on T1-W images and high to low signal intensity on T2-W images. Hypointensity on T2-W images has been shown to be caused by a greater collagenous component and reduced cellularity compared with lesions showing high signal intensity [6, 8]. Myofibromatosis is characterized by solitary or multiple lesions involving skin, subcutaneous tissues, muscle, bones and viscera. The tumors occasionally contain a central focus of hemorrhage, cystic degeneration, necrosis, calcification, ossification and fat, which explains the target sign appearance on US and MRI [6].
Fibrous hamartoma of infancy has specific histologic features. There are different triphasic mesenchymal tissues with variable relative proportions. Microscopically, fibrous hamartoma of infancy is formed of fibroblasts, myofibroblasts, primitive mesenchymal cells, small blood vessels, and mature adipocytes [6, 7]. Our patient also showed the characteristic components of fibrous hamartoma of infancy, including well-formed fibrous trabecules that were composed of dense fibrocollagenous tissue and mature adipose tissue, which were haphazardly admixed between fibrous trabeculae. Moreover, as in our patient, TS is prone to give rise to hamartomas. Therefore, based upon the appropriate clinical and histologic findings, we believe that our case corresponds to fibrous hamartoma of infancy.
In summary, although fibrous hamartoma of infancy is rare, it should be considered in the differential diagnosis of a soft-tissue mass arising in the abdominal wall in a child with TS.
References
Narayanan V (2003) Tuberous sclerosis complex: genetics to pathogenesis. Pediatr Neurol 29:404–409
Webb DW, Clarke A, Fryer A et al (1996) The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol 135:1–5
Lee JT, Girvan DP, Armstrong RF (1998) Fibrous hamartoma of infancy. J Pediatr Surg 23:759–761
Niida Y, Stemmer-Rachamimov AO, Logrip M et al (2001) Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 69:493–503
Renuka L, Thomas K, Jeanna W (2005) Fibrous hamartoma of infancy: a case report with associated cytogenetic findings. Arch Pathol Lab Med 129:520–522
Eich GF, Hoeffel JC, Tschäppeler H et al (1998) Fibrous tumors in children: imaging features of a heterogeneous group of disorders. Pediatr Radiol 28:500–509
Loyer EM, Shabb NS, Mahon TG et al (1992) Fibrous hamartoma of infancy: MR-pathologic correlation. J Comput Assist Tomogr 16:311–313
Ahn JM, Yoon HK, Suh YL et al (2000) Infantile fibromatosis in childhood: findings on MR imaging and pathologic correlation. Clin Radiol 55:19–24
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Han, HJ., Lim, GY. & You, CY. A large infiltrating fibrous hamartoma of infancy in the abdominal wall with rare associated tuberous sclerosis. Pediatr Radiol 39, 743–746 (2009). https://doi.org/10.1007/s00247-009-1218-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-009-1218-5