
Abstract
Primary atypical teratoid/rhabdoid tumor (AT/RhT) of the central nervous system is a recently described, highly malignant neoplasm in infants and young children. This tumor is an unusual combination of mixed cellular elements, similar but not typical of teratomas, and rhabdoid cells. This tumor is most common in the posterior fossa in children less than 2 years, and is radiologically similar to medulloblastoma. No pathognomonic imaging features are present. The two tumors can be separated on histologic, molecular, and cytogenetic grounds. Separation of these two tumor types is crucial because the prognosis for AT/RhT is grim even with current multimodality treatment. We present four consecutive cases of AT/RhT, three in locations other than the cerebellum, seen at our institution in a 14-month period, indicating that this tumor may be more common than previously thought.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The most common biologically malignant central nervous system (CNS) tumor occurring during the first decade of life is a primitive neuroectodermal tumor (PNET). These typically arise in the cerebellum and are known as medulloblastoma (MB). Over the past 10 years it has become apparent that a tumor with somewhat similar histological but dramatically different biological features has been misdiagnosed by many pathologists as a PNET-MB. This unique, biologically aggressive central nervous system neoplasm is formed wholly or partly by rhabdoid cells, areas resembling typical PNET, and malignant mesenchymal and/or epithelial tissue. The tumor has been named atypical teratoid/rhabdoid tumor (AT/RhT) and is regarded as a unique class of primary CNS tumors [1]. It occurs most commonly in children less than 2 years of age, has often metastasized throughout the CNS at presentation, does not respond well to therapy, and causes death usually within a year after diagnosis. These tumors may occur in any CNS location, most commonly the cerebellum (60%) [2]. Although indistinguishable grossly and radiologically, study of the microscopic features by routine and special techniques allows separation of classic PNET-MB from AT/RhT [3].
Case reports
Case 1
A 3-year-old white girl presented with 1 month of headache, vomiting, and fever, as well as a few days of head tilt, slurred speech, and difficulty walking. Upon transfer, she was lethargic with bradycardia and stiff neck. Brain and spine MRI showed a fourth ventricle mass with drop metastases involving the spinal cord, a pattern typically seen with medulloblastoma (Fig. 1). Craniotomy showed dural involvement with tumor; biopsy revealed AT/RhT. She was treated with chemo- and radiotherapy with slow clinical and radiologic improvement. Four months after diagnosis there was marked clinical deterioration with metastatic tumor by MRI. Terminal care was arranged and she died 2 weeks later.

Case 1: Gadolinium-enhanced sagittal T1-weighted images of the brain (a) and spine (b) show an enhancing mass within and expanding the fourth ventricle with sugar coating leptomeningeal enhancement along the spinal cord, mimicking medulloblastoma
Case 2
A 4-year-old white boy presented with 3–4 weeks of disordered eye movements, Parinaud's syndrome (paralysis of upward gaze), and occasional emesis. Brain and spine MRI showed a pineal mass (Fig. 2) with obstructive hydrocephalus without intracranial or spinal metastases; CSF was negative for tumor. Biopsy revealed AT/RhT. He was treated with chemotherapy and peripheral stem cell harvest with intent for radiotherapy. MRI following chemotherapy showed new intracranial metastatic tumor; CSF was positive for tumor. A parental decision was made to abandon therapy, with death 2 months after diagnosis.

Case 2: Gadolinium-enhanced sagittal and coronal T1-weighted images of the brain show a lobulated heterogeneously enhancing pineal mass with mass effect upon the tectum and obstructive hydrocephalus
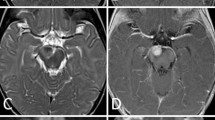
Case 3
A 5-year-old Hispanic girl presented with 1-week history of headache and vomiting. Physical examination showed papilledema. Brain and spine MRI showed a hypothalamic septal mass (Fig. 3) with obstructive hydrocephalus without intracranial or spinal metastases. Ventriculoscopic biopsy revealed AT/RhT and a ventriculoperitoneal shunt was placed. Postoperative course was complicated by panhypoituitarism and severe diabetes insipidus. She was treated with chemo- and radiotherapy with prolonged marrow hypoplasia. MRI showed a decrease in the size of the hypothalamic mass. She was taken off therapy and is in neurologically good condition 1.5 years after diagnosis.

Case 3: Axial T2 (a) and gadolinium-enhanced sagittal T1-weighted (b) images of the brain show a heterogeneous hypothalamic septal high-signal T2 mass with little enhancement and obstructive hydrocephalus
Case 4
A 4-month-old white boy presented with lethargy, vomiting, and fussiness. A left VI nerve palsy was noted and poor vision was suspected. MRI showed a large right thalamic mass extending to the midbrain (Fig. 4). Partial resection was performed, and pathology revealed AT/RhT, confirmed on review at the Mayo Clinic. The postoperative course was complicated by subdural hematomas and hydrocephalus. MRI performed 10 days after partial resection showed rapid regrowth of the tumor. Chemotherapy was given with a decrease in the size of the mass. His clinical condition rapidly deteriorated over the next week, and imaging again showed rapid regrowth of the tumor. He was placed on terminal care and died 6 weeks after presentation.

Case 4: Axial (a) and sagittal (b) gadolinium-enhanced T1-weighted images of the brain show an enhancing midline mass arising from the right thalamus and extending to the midbrain
Discussion
Malignant rhabdoid tumors are biologically aggressive neoplasms that occur most frequently in the kidneys of infants and children. Similar tumors occur in other locations, including the central nervous system. In the brain, they may be composed purely of rhabdoid cells (malignant rhabdoid tumor, MRT), or have a mixture of rhabdoid cells, neuroepithelial, epithelial, and mesenchymal elements [4]. Because of the multiplicity of cell types in this tumor, it has been likened to a type of teratoma. The tumor is histologically different from more familiar teratomas, and is negative for routine germ-cell markers. The capacity for divergent differentiation and the presence of rhabdoid cells led Rorke et al. to coin "atypical teratoid/rhabdoid tumor" (AT/RhT) as a descriptive diagnostic term [1]. AT/RhTs occur most commonly in the posterior fossa and have been mistaken for medulloblastoma. Such confusion may account for the perception that infants with PNET-MB who are 2 years or younger have a worse prognosis than do other children with such tumors.
Two of the four children in our series were 3 years or younger at diagnosis; however, the other two were older. Only one child's tumor was in a typical posterior fossa location, with the others in an atypical supratentorial location. In a multicenter study of 52 infants and children with AT/RhT [3], ¾ of patients were aged 3 years or younger at the time of diagnosis (median age of 16.5 months). AT/RhT tumors were infratentorial in 33 patients (29 cerebellum, 4 cerebellopontine angle) and supratentorial in 14 (3 pineal, 10 cerebral hemisphere, 1 suprasellar, 1 extramedullary spinal cord) and multifocal in 4. Children younger than 3 years often presented with nonspecific signs such as lethargy, vomiting, head tilt and failure to thrive. Children with cerebral AT/RhT are more likely to present at an older age, as in two of the four children in our series.
The radiologic features of AT/RhT are nonspecific, as in the four cases presented, but may be similar to PNET-MB. There is increased density on nonenhanced CT and heterogeneous contrast enhancement. Cysts and hemorrhage are common. With MRI there is decreased signal intensity on T1- weighted images, iso- or decreased density on T2-weighted images (due to hypercellularity), and heterogeneous enhancement post gadolinium [5]. Therefore, the radiological differential diagnosis before histochemistry analysis includes PNET-MB, teratoma, astrocytoma, choroid plexus papilloma, and ependymoma.
Approximately one-third of patients with AT/RhT have intracranial dissemination with involvement of the cerebrospinal fluid (CSF) at diagnosis and two-thirds at relapse [3]. Two of the four children in the current series had CSF dissemination of tumor. The most consistent cytologic feature of AT/RhT are the large rhabdoid cells [6]. Cytological analysis of the CSF is important in determining the extent of tumor involvement and in monitoring the response to therapy. There is one report of peritoneal shunt metastses in a child with an AT/RhT [7].
AT/RhTs exhibit variations in cell type and histologic pattern. Immunohistochemical investigation is most helpful in diagnosis. The three antibodies of greatest value are vimentin, epithelial membrane antigen (EMA), and smooth muscle actin (SMA). The rhabdoid cells almost always express EMA, vimentin, and SMA in various degrees [8]. Although AT/RhT of the CNS has been characterized as a clinicopathologic entity, the histogenesis remains unknown [1]. The major cytologic differential diagnosis of AT/RhT is medulloblastoma. AT/RhT often includes cellular areas with histologic components consistent with primitive neuroectodermal tumors. Tumor cells of medulloblastoma are relatively small, with scanty cytoplasm and hyperchromatic nuclei. Large rhabdoid cells are not found in medulloblastoma [6]. Cytogenetic studies of AT/RhTs reveal monosomy and various translocations and deletions of chromosome 22 in greater than 50% of cases [2]. Several patients with AT/RhTs have cytogenetically normal chromosome 22 and complex rearrangements of chromosomes 6 and 11 [4]. The disparity in cytogenetic data probably reflects clinical and morphological heterogeneity of tumors sharing the rhabdoid phenotype. Anormalities of chromosome 22 distinguish AT/RhTs from PNETs that typically display an i(17q) abnormality [3].
It is suspected that AT/RhTs have been hidden among PNET-MB in the past . A systematic review of 127 surgical specimens diagnosed as PNET-MB in a 23-year period was undertaken to estimate the frequency of AT/RhT [9]. In addition to 5 originally diagnosed AT/RhTs at the same institution in the same time period, 6 cases were reclassified as AT/RhT. Ten of these 11 cases were located in the cerebellum and 1 in the cerebral hemisphere. The sole feature in common was the presence of large, pale, bland rhabdoid cells, some with a sickle or embracing shape. The frequency of AT/RhT expressed as a ratio of AT/RhT to PNET-MB was 1:11 in general and increased to 1:3.8 among patients younger than 3 years of age. There was a shorter survival time in patients with AT/RhT compared to PNET-MB: 15.4 months versus 156.4 months, respectively [9].
Mean survival time is has been reported as 3 months after only surgical intervention and 8 months with adjuvant chemotherapy and radiotherapy. The majority of patients with AT/RhTs have died within 1 year of diagnosis, in contrast to children with PNET-MB, among whom there is at least a transient response to chemotherapy in 40–60% [3]. Standard therapy for infant and childhood medulloblastoma has been ineffective for AT/RhT [10]. Intensified therapy has been recently used with promising results. In a report of four cases of AT/RhT in young children with subtotal resection and localized disease at diagnosis, two children who received high-dose chemotherapy and autologous bone-marrow transplant had a good response (one survived 19 months and the other is free of disease 46 months from diagnosis) [11]. Therefore, intensified therapy may alter the natural history of AT/RhT.
In summary, CNS AT/RhT is a distinct, highly malignant neoplasm that occurs in infants and young children, is defined by the presence of rhabdoid cells and heterogeneity of cell type, and is associated with deletion of chromosome 22. It most commonly occurs in the posterior fossa, mimicking medulloblastoma, but can occur in the supratentorial compartment. One-third of children have CSF dissemination at diagnosis. The prognosis has been dismal with future hope using intensified therapy. This tumor should be considered in the differential diagnosis for infants with radiologic findings mimicking medulloblastoma and in children with a supratentorial mass, especially if there is CSF dissemination.
References
Lefkowitz IB, Rorke LB, Packer RJ, et al (1987) Atypical teratoid tumor of infancy: definition of an entity. Ann Neurol 22:448–449
Rorke LB, Packer RJ, Biegel JA (1995) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 24:21–28
Rorke LB, Packer RJ, Biegel JA (1996) Central nervous system atypical teratoid/ rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85:56–65
Lopez-Gines C, Cerda-Nicolas M, Kepes J, et al (2000) Complex rearrangement of chromosomes 6 and 11 as the sole anomaly in atypical teratoid/rhabdoid tumors of the central nervous system. Cancer Genet Cytogenet 122:149–152
Zuccoli G, Izzi G, Bacchini E, et al (1999) Central nervous system atypical teratoid/rhabdoid tumour of infancy: CT and MR findings. Clin Imaging 23: 356–360
Lu L, Wilkinson EJ, Yachnis AT (2000) CSF cytology of atypical teratoid/rhabdoid tumor of the brain in a two-year-old girl: a case report. Diagn Cytopathol 23:329–332
Korones DN, Meyers SP, Rubio A, et al (1999) Brief report: a 4-year-old girl with a ventriculoperitoneal shunt metastasis of a central nervous system atypical teratoid/rhabdoid tumor. Med Pediatr Oncol 32:389–391
Hauser P, Slowik F, Bognar L, et al (2001) Atypical teratoid/rhabdoid tumor or medulloblastoma? Med Pediatr Oncol 36:644–648
Ming-Tak Ho D, Hsu CY, Wong TT, et al (2000) Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99:482–488
Burger PC, Yu IT, Tihan T, et al (1998) Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a pediatric oncology study. Am J Surg Pathol 22:1083–1092
Hilden JM, Watterson J, Longee DC, et al (1998) Central nervous system atypical teratoid tumor/rhabdoid tumor: response to intensive therapy and review of the literature. J Neurooncol 40:265–275
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fenton, L.Z., Foreman, N.K. Atypical teratoid/rhabdoid tumor of the central nervous system in children: an atypical series and review. Pediatr Radiol 33, 554–558 (2003). https://doi.org/10.1007/s00247-003-0934-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-003-0934-5