
Abstract
Left ventricular noncompaction (LVNC) is a genetically heterogeneous disorder the etiology of which is still debated. During fetal development, trabecular cardiomyocytes contribute extensively to the working myocardium and the ventricular conduction system. The impact of developmental defects in trabecular myocardium in the etiology of LVNC has been debated. Recently we generated new mouse models of LVNC by the conditional deletion of the key cardiac transcription factor encoding gene Nkx2-5 in trabecular myocardium at critical steps of trabecular development. These conditional mutant mice recapitulate pathological features similar to those observed in LVNC patients, including a hypertrabeculated left ventricle with deep endocardial recesses, subendocardial fibrosis, conduction defects, strain defects, and progressive heart failure. After discussing recent findings describing the respective contribution of trabecular and compact myocardium during ventricular morphogenesis, this review will focus on new data reflecting the link between trabecular development and LVNC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Left ventricular noncompaction (LVNC) is a complex and heterogeneous cardiomyopathy, characterized by hypertrabeculation and deep trabecular recesses in the left ventricle and is the most common cardiomyopathy with a large spectrum of symptoms ranging from normal variants to a pathological phenotype [1]. Patients display a high prevalence of conduction defects. The etiology of LVNC is still debated within the scientific and medical community, in particular the impact of developmental defects of ventricular trabeculae on the occurrence of LVNC [2]. Ventricular trabeculation is a normal, but transient, step during embryonic development [3, 4]. In mammals, trabeculae undergo a necessary compaction step at fetal stages during formation of a competent myocardial wall. Trabeculae also contain the progenitor cells of the ventricular conduction system (VCS), controlling the rapid propagation of the electrical activity in the ventricles [5, 6]. Recently, it has been demonstrated that embryonic trabeculae play an important role in the formation of the ventricular wall including the VCS, and that failure of their development triggers pathological features of LVNC similar to those observed in symptomatic patients [7]. In this review, we focus on the development and cell fate of ventricular trabeculae, highlighting their potential role in cardiac diseases.
Ventricular Trabeculae
Trabeculae are transient embryonic structures forming finger-like myocardial projections in the lumen of ventricles, giving the heart a sponge like structure [8, 9]. They play a role in oxygenating the myocardium prior to the establishment of the coronary vascularization, and a role in fast conduction by expressing the gap junction protein Connexin40 (Cx40) before the development of the ventricular conduction system (VCS) [10, 11]. Trabeculae are apparent at embryonic day (E) 9.0 in the mouse or at the end of the fourth gestational week in human, when the heart tube consists of an external myocardial layer and an internal endocardial epithelium. These two layers are separated by extracellular matrix known as the cardiac jelly. The first trabeculae appear in the cardiac jelly between endocardial-myocardial contact points and extend radially into the ventricular lumen at the outer curvature of the looped-heart tube [10]. This process, known as trabeculation, occurs in all vertebrates but diverges between species. Here we discuss mechanisms that have been recently uncovered that provide insight into the molecular and cellular pathways driving trabeculae formation.
Morphogenesis of Embryonic Trabeculae in Mammals
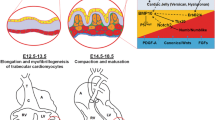
Trabeculation depends on tissue mechanics, cardiac jelly composition, cell behavior, and cell-to-cell signaling between the endocardium and the myocardium [12,13,14]. In mammals, both oriented cell division and delamination of cardiomyocytes from the early myocardial layer are important early events in the formation of trabeculae. In the mouse, lineage tracing of single cells during trabeculation revealed that, in addition to polarized migration similar to that observed in zebrafish [15], cardiomyocytes undergo perpendicular cell division at E9.0/9.5, leading to the entry of a daughter cell into the cardiac lumen to initiate trabeculation. In this way the two daughter cells exhibit different localizations in the ventricle and are likely to respond to different developmental cues leading to their segregation [16]. During this process N-cadherin reorganization is required for the establishment of mitotic spindle orientation in order to drive cardiomyocytes delamination and perpendicular division [16].
Recently a new model of trabeculation in mice has been proposed that integrates dynamic endocardial and myocardial cell behaviors with ECM remodeling [17]. The first apparition of trabeculae occurs at E8.0 in mouse embryo, when several endocardial sprouts ingress though the cardiac jelly and toward the myocardium. At this stage, the myocardium consists of a continuous layer of cardiomyocytes at the most outer ventricular surface and discontinuous luminal cardiomyocyte clusters defined as trabecular units. From E8.5 to E9.0, endocardial projections encapsulate these trabecular units and establish points of contact with the compact myocardium. This process individualizes trabecular units and creates multiple matrix rich areas. From E9.0, trabeculae continue to expand by radially orientated cardiomyocyte proliferation and potentially by recruitment of new cardiomyocytes until the end of ventricular septation at E14.5 in mouse embryos. By this stage, the ventricular endocardium becomes tightly associated with the myocardial cells as the intervening cardiac jelly disappears. The trabecular zone constitutes the most important mass of the ventricular myocardium, whereas the outer compact layer remains only a few cells thick. During trabeculation, the endocardial Neuregulin 1 and NOTCH1 pathways cooperate dynamically to regulate the balance between ECM synthesis and degradation, essential for trabecular architecture and growth [17], and also regulate cardiomyocyte proliferation by promoting the expression of Bmp10 [18]. Furthermore, the ECM composition during trabecular growth is dynamically controlled by the secreted matrix metalloproteinase ADAMTS1 and its chromatin-based transcriptional repressor Brg1 [19]. In mice mutant for genes in these pathways, higher ECM degradation blocks trabecular growth, whereas the absence of ECM degradation leads to excessive and disorganized trabecular growth contributing to noncompaction cardiomyopathy phenotypes. Nevertheless, how endocardial sprouting is regulated and the cellular and molecular mechanisms that drive the formation of trabecular units remain to be elucidated.
Compact Versus Trabecular Cardiomyocytes
Ventricular trabeculation generates two myocardial domains: a trabecular zone adjacent to the endocardium and a sub-epicardial compact zone [4]. Lineage analyses in chick and mouse embryos have shown that compact and trabecular cardiomyocytes are clonally related [20, 21]. However they differ in term of morphogenesis, proliferative capacities, gene expression and contribution to the adult cardiac structures [3]. There is a gradient of cardiomyocyte proliferation across the ventricular wall. Trabecular cardiomyocytes have a lower proliferative rate than those in the compact wall. Indeed, a balance between proliferation and differentiation is critical to both generate a sufficient number of cells and initiate lineage specification [22]. The trabecular zone is characterized by the expression of the genes Irx3, Cx40, Nppa, Etv1, Bmp10, Slit2, and Sema3a whereas the compact zone is characterized by the expression of Tbx20, Hey2, Loxl2, and N-Myc [6, 22,23,24,25,26,27,28] (Fig. 1a).

Molecular identity and genetic tracing of trabecular cells. a Spatiotemporal expression pattern of trabecular markers and compact zone markers in the ventricular myocardium during development. Trabecular markers are downregulated in the compact zone at E14.5 but maintained in trabecular cells (orange shading) that differentiate into contractile and conductive cardiomyocytes. Working cardiomyocytes deriving from trabeculae progressively lose the expression of trabecular markers (black dots). At the adult stage, the expression of trabecular markers Cx40, Sema3a, Irx3, and Etv1 is restricted to Purkinje fibers (PF) expressing also HCN4 and CNTN2. b Genetic tracing of trabecular cells in adult mice showing the distribution of trabecular-derived cells (green dots) in transverse sections at the mid ventricle after genetic labeling at E10.5 or E14.5. Labeling at E10.5 shows large contribution of trabecular cells to the working myocardium of the left ventricle (LV) extended toward the compact myocardium, including the left free wall and the papillary muscles. Labeling at E14.5 shows a more restricted distribution in the inner part of the myocardium at a subendocardial level. Labeling at E18.5 shows the restriction of trabecular cells into the PF (red)
Cell Fate of Trabeculae During Ventricular Wall Formation in Mammals
Trabecular Compaction
In mammals, from fetal stages trabeculae are gradually compacted and integrated into the ventricular wall thickness, concomitant with the expansion of the compact zones [3, 4, 29]. This results in the formation of a thick and competent myocardial wall. The compaction step is specific to higher vertebrates since ventricular trabeculae are maintained in the heart of adult fishes and reptiles which retain regenerative capacity [9, 30]. In adult zebrafish heart, tbx5a expression is persistent only in the trabecular myocardium and genetic fate mapping of trabecular-tbx5a+ cells shows that these cells are able to switch their fate and differentiate into cortical myocardium during both normal development and adult heart regeneration [31]. This suggests that trabecular myocardium displays a high degree of cell plasticity and/or retains proliferative ability.
From E14.5 of mouse development, trabeculae stop extending and start to consolidate at their base and gradually integrate into the ventricular wall thickness, a process that is complete by early postnatal stages. The thickening of the myocardial wall starts from the base of the ventricle and progresses towards the apex. Finally the trabecular projections disappear as the compact zone grows and maturates, resulting in a ventricular wall that is largely composed of compact myocardium with a relatively smooth endocardial surface [4]. During ventricular compaction, cardiomyocytes of the trabecular myocardium participate in the formation of the papillary muscles and VCS, whereas endocardial cells associated with the base of trabeculae contribute to the coronary vasculature. In fact during compaction the intertrabecular recesses are compressed to form capillaries and endothelial cells arising from the endocardium contribute to intramyocardial coronary vessels [32, 33]. A similar process takes place during neo-vascularization following myocardial infarction in the adult heart [34]. Epicardial-derived cells and cells from the sinus venosus also participate in building the coronary vasculature [35, 36].
The process of compaction is poorly described in the literature and often causes confusion. Certain authors support a dynamic coalescence of ventricular trabeculae, while others argue in favor of a remodeling of the compact zone that undergoes an extensive proliferation resulting in the zipping-up of trabeculae [24]. Interestingly, recent genetic tracing and retrospective clonal analysis in the mouse has suggested that the compaction step may be in part driven by expansion of the fetal compact myocardium into the trabecular layer through its higher proliferative activity. This leads to the formation of a hybrid zone composed of cardiomyocytes derived from both the compact and trabecular zones [24]. Preventing proliferation of cardiomyocytes in the compact zone compromises the formation of the hybrid zone, and results in a thin and non-compacted ventricular myocardium with persistent trabeculae associated with contraction defects. In contrary, preventing proliferation in the trabecular zone does not perturb ventricular compaction. These results are consistent with the higher proliferative activity of the compact zone and the switch from trabecular expansion to compact zone expansion during the compaction step. Nevertheless, this study does not exclude that both expansion of the compact zone and coalescence of intertrabecular recesses are two critical events that drive the compaction process.
Trabecular Specification into Conductive and Contractile Cardiomyocytes
As trabeculae gradually integrate the compact wall, trabecular cardiomyocytes differentiate into contractile, also designed as working, cardiomyocytes and conductive cells. Lineage tracing experiments and retrospective clonal analysis of Cx40+ cells have demonstrated that the peripheral VCS including Purkinje fibers (PFs), originates from embryonic trabeculae [5]. Ventricular trabeculae contain bipotent myogenic progenitors which segregate progressively into conductive and contractile fates between E10.5 and E14.5; lineage restriction appears almost complete by E16.5. Segregation into a conductive fate is associated with a reduced proliferative capacity compared to contractile cardiomyocytes that exhibit a high proliferative rate until perinatal stages [37]. Furthermore, the progressive segregation of trabecular cardiomyocytes into cells with a contractile myocardial fate is accompanied by the loss of expression of trabecular markers including Etv1, Irx3, Cx40, and Sema3a, the expression of which persists in the VCS at postnatal stages [6, 23, 25, 26, 38] (Fig. 1b).
Indeed, ventricular wall morphogenesis seems to result from a continuous process during which trabecular cells are progressively incorporated into the compact myocardium. This process follows the formation of the ventricular wall from base to apex as demonstrated by the more severe defects observed in the apex region in mutant mice or in human patients suffering from LVNC [39, 40]. Mutations in the NKX2-5 gene have been identified in LVNC patients indicating a role for this important cardiac transcription factor in compaction [41, 42]. In Nkx2-5 heterozygous mutant mice, noncompaction of the ventricular myocardium is observed in the apex suggesting a late compaction of the apical part, as previously suggested for the development of the coronary vasculature [36]. A similar phenotype is observed in 14-3-3ε−/− mice that display abnormal coronary vasculature and ventricular noncompaction in the apical region of the heart [43]. These observations, in addition to the early segregation of the central VCS components at the crest of the IVS [44], suggest that ventricular myocardial morphogenesis and VCS development follow the same base to apex axis pattern through embryonic development. This assumption is supported by optical mapping data showing that electrical activity propagates through a base to apex axis in early stages of embryonic development [10, 45].
LVNC: A Poorly Understood Cardiomyopathy
Left ventricular noncompaction (LVNC) is a rare genetic or acquired cardiomyopathy also known as ventricular hypertrabeculation. LVNC is typically characterized by the presence of an excessive lace-like network of trabeculae with deep intertrabecular recesses. However, the anatomical and clinical features are very heterogeneous because the degree of hypertrabeculation is highly variable among patients. Moreover, LVNC occurs in association with several genetic mutations and is often associated with other cardiac diseases, in particular dilated cardiomyopathy [1]. Thus, a large spectrum of clinical conditions varying from asymptomatic to severe pathological forms through to sudden cardiac death are observed. The most prevalent symptoms are conduction defects present in approximately 90% of patients. Around 60% of adult patients develop heart failure and around 13–24% thromboembolism [46]. It is still unclear if LVNC is a separate cardiomyopathy, or an anatomical trait observed in association with other primary cardiomyopathies. Moreover, several asymptomatic adults displaying a normal ejection fraction and no risks of complications such as arrhythmia and stroke have been observed with excessive ventricular trabeculae [2]. Consequently, LVNC prognosis and diagnosis are difficult and result in an underestimated prevalence among the worldwide population. Patients are usually diagnosed when the conditions become symptomatic or when complications occur. One study has estimated a prevalence of 9.2% among children affected by cardiomyopathies. In adults referred for echocardiography, the prevalence is 0.01–0.30% [46].
In congenital forms of LVNC, more than 40 mutated genes have been associated with noncompaction [46]. Most of these genes are also associated with other cardiomyopathies and encode for sarcomeric proteins, related binding proteins, and cytoskeletal proteins [47]. However, the evidence of a direct causative link between any of these mutations and LVNC remains to be established, illustrating the importance of generating mouse models of LVNC in order to investigate the developmental origin of this misunderstood cardiomyopathy. One challenging point concerns the etiology of LVNC which is largely unknown. It is still unclear whether hypertrabeculation results from excessive trabeculae formation and/or a defect in the later compaction processes during ventricular myocardium morphogenesis. Comparative studies, however, support an arrest of myocardial compaction during fetal stages [48]. Several studies using mouse models have argued that the phenotype of ventricular hypertrabeculation as observed in FKBP12 [49], MIB1 [50], Numb [51] and Nkx2-5 [52] mutant mice, mostly result from defective cell proliferation and are mainly associated with dysregulation in the NOTCH pathway. Others have supported that defects in cardiomyocyte myofibrillogenesis and polarization during ventricular development identify a common pathogenic pathway of LVNC [53, 54]. However, recent evidence indicates that excessive trabeculation results from defective compaction zone expansion and not from failure of the compaction of pre-existing trabeculae [2, 55]. Inhibiting proliferation of the compact zone by deleting Yap1, a key transcription co-factor required for normal cardiomyocyte proliferation, prevents the expansion of the compact zone and results in noncompaction of the ventricular myocardium [24]. Several other mouse models including the ventricular Nkx2-5-conditional deletion support this assumption; however, most of these mutants present a very thin compact layer and are embryonic lethal and thus do not fully recapitulate symptomatic features observed in LVNC patients.
Clues from Nkx2-5-Trabecular Deficient Mice Support a Developmental Origin of Pathological LVNC
In a recent study, we wanted to directly address the question whether hypertrabeculated myocardium could result from a failure of trabecular development. Defects in ventricular compaction and conduction are common traits observed in patients and in mutant mice carrying mutations in NKX2-5 gene, encoding a key transcriptional regulator of cardiac development [52, 56,57,58]. In order to study the temporal and spatial requirement of Nkx2-5 during trabecular morphogenesis and dissect the role of this gene in the pathogenesis of LVNC, we conditionally inactivated Nkx2-5 in early or late trabecular myocardium, at timepoints corresponding to trabecular morphogenesis and compaction [7]. Nkx2-5-floxed mice were crossed with the Cx40- CreERT2 mouse line, in which tamoxifen inducible Cre activity is under the transcriptional control of the Cx40 locus. In Nkx2-5ΔTrabE10 mutant mice, Nkx2-5 was deleted at the embryonic stages E10.5-E11.5, when Cx40 is expressed in developing trabeculae during active trabeculation. In Nkx2-5ΔTrabE14 mutant mice, Nkx2-5 was deleted at fetal stages E13.5−14.5, when Cx40 expression is restricted to trabeculae during their compaction.
Nkx2-5ΔTrabE10 and Nkx2-5ΔTrabE14 adult mutant mice exhibit a hypertrabeculation phenotype with deep intertrabecular recesses associated with subendocardial fibrosis and hypoplasia of the Purkinje fibers (PF), although the compact zone was not reduced. The phenotypes after early and late Nkx2-5 deletion vary only in their degree of severity. These results demonstrate that disruption of trabecular development contributes to the occurrence of hypertrabeculation (Fig. 2a). By performing inducible lineage tracing of Cx40+ trabeculae, we have demonstrated that Cx40+ cells labeled at E10-11 or E13-14, contribute predominantly to generate working cardiomyocytes that extend throughout the ventricular myocardium. Cardiomyocytes generated by Cx40+ cells labeled at E10-11 are observed deep in the myocardium, while Cx40+ cells labeled at E13-14 contribute to cardiomyocytes that are restricted to the inner myocardial layer (Fig. 1b). The difference of severity in the hypertrabeculated phenotype observed between early and late Nkx2-5-deletion is thus correlated with the extent of deletion in the ventricular myocardium rather than resulting from different mechanisms acting independently during trabeculation or trabecular compaction. Nkx2-5ΔTrabE10 mice displayed defects in papillary muscle compaction, severe PF hypoplasia, numerous endocardial islets, important subendocardial fibrosis and disruption of the molecular boundary between the compact and the trabecular zone in the adult heart. The main difference with previous models is that conditional deletion restricted to the trabecular compartment provokes a less severe phenotype and allows the development of a compact myocardium thick enough for supporting cardiac function and compatible with life. In mutant embryos, hypertrabeculation was associated with a slight upregulation of cardiac proliferation at E14.5 without disturbing the compact-trabecular boundary at this stage. The maintenance of this embryonic compact-trabecular boundary has been observed in other mouse models of noncompaction affecting notch signaling. The glycosyltransferase manic fringe (MFng) is a modulator of Notch ligand in ventricular endocardial cells and is expressed from E8.5 and downregulated after E11.5 in mouse embryos. MFngtg::Tie2-Cre transgenic mice display forced MFng expression in endocardial cells leading to disruption of the Notch1 activation in ventricular myocardium [59]. Neonatal mice exhibit thin compact myocardium and non-compacted trabeculae in both ventricles. In E16.5 mutant embryos, the boundary between compact and trabecular myocardium is maintained since Hey2 and Bmp10 (or Cx40) expressions remain complementary, despite the presence of deep recesses in the compact myocardium referred as the “intermediate myocardium”.

[7]
Mouse models of pathological LVNC. a Short-axis cine images recorded by magnetic resonance imaging at end-diastole (upper panels). Anatomical structure of opened left ventricle (LV) (mid panels). Immunostaining on traverse sectioned-mid left ventricle of 3 month-old control (CTL), Nkx2-5ΔTrabE10 (ΔTrbE10) and Nkx2-5ΔTrabE14 (ΔTrbE14) show the morphology of adult hypertrabeculation and deep intertrabecular recesses (white arrows) induced by deletion of Nkx2-5 in trabecular cardiomyocytes during development. b The follow-up of cardiac function shows a correlation of impaired Ejection Fraction with conduction and strain defects in absence of aggravation of the hypertrabeculation phenotype with age. HF: Heart failure. IVS: Interventricular septum. *Papillary muscles. Adapted from Choquet et al.
Finally our results suggest that hypertrabeculation phenotype results primarily from both early cardiac proliferation defects and impaired trabecular coalescence. Thus, trabecular cells are actively involved in the process of compaction. Moreover, the presence of deep endocardial invaginations and transcriptional deregulation of numerous genes involved in the vascular system suggest that ventricular compaction involves an intimate communication between cardiomyocytes and endothelial cells. However, the absence of defects in coronary arteries in contrast to other models shows that noncompaction can be dissociated from coronary artery development. Furthermore, transcriptomic analysis demonstrates no differences in dysregulated pathways between Nkx2-5ΔTrabE10 and Nkx2-5ΔTrabE14 adult mutant mice. This suggests that Nkx2-5 regulates common developmental processes throughout trabecular development. While previous data favor defects in proliferation of the compact myocardium, our results provide evidence that defects in trabecular morphogenesis also lead to mouse models of LVNC and suggest that trabecular compaction and compact zone expansion are progressive processes.
In the first longitudinal study of a LVNC model we followed-up cardiac function in conditional Nkx2-5 mutant mice over one year. Our results revealed that Nkx2-5ΔTrabE10 and Nkx2-5ΔTrabE14 mutant mice display all the clinical signs of symptomatic LVNC, including conduction defects, progressive cardiac contractility defects with age and in 50% of old mutant mice signs of heart failure, although, importantly, the hypertrabeculation and fibrosis phenotypes remain identical throughout adult life (Fig. 2b). Thus, these mutant mice provide good models for studying LVNC and highlight the multiple roles of Nkx2-5, reflecting the heterogeneity of clinical features observed in LVNC patients. Finally, our results support the idea that even if excessive trabeculation is not itself a clinical entity, LVNC may be attributed to a defect in the global and progressive maturation of the ventricular myocardium that could be the primary cause of the LVNC pathology.
Future Directions
Consistent with the intimate relationship between VCS and ventricular wall formation, the maturation state of trabeculae is an important criterion to take in consideration regarding LVNC cardiomyopathy, and not exclusively the persistence of excessive trabeculation in the ventricular cavity. In this case the failure of trabecular cardiomyocytes to adopt a conductive fate would impact ventricular wall development, especially the formation of the VCS and the appearance of subendocardial fibrosis and intertrabecular recesses. Lineage tracing of Cx40+ trabecular cells indicates that the coalescence of trabeculae into the ventricular wall correlates with the progressive restriction of Cx40 expression to the conduction system. This pattern of expression is similar to that of Sema3a, another PF marker [26]. Furthermore, this lineage restriction is accompanied by a decrease in the number of Cx40+ trabecular progenitors, and a progressive integration of Cx40-derived cells into the working myocardium. This demonstrates that trabecular development is concomitant with VCS differentiation and that PF hypoplasia is likely to be directly linked to the conduction defects observed in our LVNC mouse models. Thus, disturbing trabecular differentiation may have an important impact in the pathogenicity of hypertrabeculation and correlates with symptomatic forms of this cardiac anomaly. In the future, it would be relevant to further study the impact of defective VCS alone to decipher its role in pathological features of cardiomyopathies.
References
Towbin JA, Lorts A, Jefferies JL (2015) Left ventricular non-compaction cardiomyopathy. Lancet 386:813–825. https://doi.org/10.1016/S0140-6736(14)61282-4
Anderson RH, Jensen B, Mohun TJ et al (2017) Key questions relating to left ventricular noncompaction cardiomyopathy: is the emperor still wearing any clothes? Can J Cardiol 33:747–757. https://doi.org/10.1016/j.cjca.2017.01.017
Samsa LA, Yang B, Liu J (2014) Embryonic cardiac chamber maturation: trabeculation, conduction and cardiomyocyte proliferation. Am J Med Genet C 1:20. https://doi.org/10.1002/ajmg.c.31366.Embryonic
Zhang W, Chen H, Qu X et al (2013) Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet Part C 163:144–156. https://doi.org/10.1002/ajmg.c.31369
Miquerol L, Moreno-Rascon N, Beyer S et al (2010) Biphasic development of the mammalian ventricular conduction system. Circ Res 107:153–161. https://doi.org/10.1161/CIRCRESAHA.110.218156
Beyer S, Kelly RG, Miquerol L (2011) Inducible Cx40-Cre expression in the cardiac conduction system and arterial endothelial cells. Genesis 49:83–91. https://doi.org/10.1002/dvg.20687
Choquet C, Nguyen THM, Sicard P et al (2018) Deletion of Nkx2-5 in trabecular myocardium reveals the developmental origins of pathological heterogeneity associated with ventricular non-compaction cardiomyopathy. PLoS Geent 14:e1007502
Challice CE, Viragh S (1974) The architectural development of the early mammalian heart. Tissue Cell 6:447–462. https://doi.org/10.1016/0040-8166(74)90037-8
Thomas PS, Sedmera D (1996) Trabeculation in the embryonic heart. Comp Gen Pharmacol 18:607
Sedmera D, Pexieder T, Vuillemin M et al (2000) Developmental patterning of the myocardium. Anat Rec 258:319–337. https://doi.org/10.1002/(SICI)1097-0185(20000401)258:4%3c319:AID-AR1%3e3.0.CO;2-O
Sankova B, Benes J, Krejci E et al (2012) The effect of connexin40 deficiency on ventricular conduction system function during development. Cardiovasc Res 95:469–479. https://doi.org/10.1093/cvr/cvs210
Cherian AV, Fukuda R, Augustine SM et al (2016) N-cadherin relocalization during cardiac trabeculation. Proc Natl Acad Sci USA 113:7569–7574. https://doi.org/10.1073/pnas.1606385113
Staudt DW, Liu J, Thorn KS et al (2014) High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development 141:585–593. https://doi.org/10.1242/dev.098632
Han P, Bloomekatz J, Ren J et al (2016) Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature 534:700–704. https://doi.org/10.1038/nature18310
Peshkovsky C, Totong R, Yelon D (2011) Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Dev Dyn 240:446–456. https://doi.org/10.1002/dvdy.22526
Li J, Miao L, Shieh D et al (2016) Single-cell lineage tracing reveals that oriented cell division contributes to trabecular morphogenesis and regional specification. Cell Rep 15:158–170. https://doi.org/10.1016/j.celrep.2016.03.012
del Monte-Nieto G, Ramialison M, Adam AAS et al (2018) Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 557:439–445. https://doi.org/10.1038/s41586-018-0110-6
Grego-bessa J, Luna-zurita L, Monte G et al (2007) Notch Signalling is essential for ventricular development. Dev Cell 12:415–429. https://doi.org/10.1016/j.devcel.2006.12.011.Notch
Stankunas K, Hang CT, Tsun Z et al (2009) Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell 14:298–311. https://doi.org/10.1016/j.devcel.2007.11.018.Endocardial
Cheng G, Litchenberg WH, Cole GJ et al (1999) Development of the cardiac conduction system involves recruitment within a multipotent cardiomyogenic lineage. Development 126:5041–5049. https://doi.org/10.1038/nature09714
Meilhac SM, Kelly RG, Rocancourt D et al (2003) A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development 130:3877–3889. https://doi.org/10.1242/dev.00580
Bossie A, Ph D, Greenspan A et al (2004) BMP-10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 16:21–30. https://doi.org/10.1097/JGP.0b013e31813546f2.Placebo-Controlled
Kim K-H, Rosen A, Hussein SMI et al (2016) Irx3 is required for postnatal maturation of the mouse ventricular conduction system. Sci Rep 6:19197. https://doi.org/10.1038/srep19197
Tian X, Li Y, He L et al (2017) Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat Commun 8:87. https://doi.org/10.1038/s41467-017-00118-1
Shekhar A, Li X, Liu F-Y et al (2016) Transcription factor ETV1 is essential for rapid conduction in the heart. J Clin Invest 1:11. https://doi.org/10.1172/JCI87968.Introduction
Li Y, Tian X, Zhao H et al (2018) Genetic targeting of Purkinje fibres by Sema3a- CreERT2. Sci Rep 8:1–9. https://doi.org/10.1038/s41598-018-20829-9
Zhang W, Chen H, Wang Y et al (2011) Tbx20 transcription factor is a downstream mediator for bone morphogenetic protein-10 in regulating cardiac ventricular wall development and function. J Biol Chem 286:36820–36829. https://doi.org/10.1074/jbc.M111.279679
Li G, Xu A, Sim S et al (2016) Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell 39:491–507. https://doi.org/10.1016/j.devcel.2016.10.014
Foglia MJ, Poss KD (2016) Building and re-building the heart by cardiomyocyte proliferation. Development 143:729–740. https://doi.org/10.1242/dev.132910
Jensen B, Wang T, Christoffels VM, Moorman AFM (2013) Evolution and development of the building plan of the vertebrate heart. Biochim Biophys Acta - Mol Cell Res 1833:783–794. https://doi.org/10.1016/j.bbamcr.2012.10.004
Sánchez-Iranzo H, Galardi-Castilla M, Minguillón C et al (2018) Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nat Commun. https://doi.org/10.1038/s41467-017-02650-6
Wu B, Zhang Z, Lui W et al (2012) Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 151:1083–1096. https://doi.org/10.1016/j.cell.2012.10.023.Endocardial
Tian X, Hu T, Zhang H et al (2014) De Novo formation of a distinct coronary vascular population in neonatal heart. Science 49:1841–1850. https://doi.org/10.1016/j.jacc.2007.01.076.White
Miquerol L, Thireau J, Bideaux P et al (2015) Endothelial plasticity drives arterial remodeling within the endocardium after myocardial infarction. Circ Res 116:1765–1771. https://doi.org/10.1161/CIRCRESAHA.116.306476
Miquerol L, Kelly RG (2012) Organogenesis of the vertebrate heart. Wiley Interdiscip Rev Dev Biol 2:17–29. https://doi.org/10.1002/wdev.68
Rhee S, Chung JI, King DA et al (2018) Endothelial deletion of Ino80 disrupts coronary angiogenesis and causes congenital heart disease. Nat Commun. https://doi.org/10.1038/s41467-017-02796-3
Sereti KI, Nguyen NB, Kamran P et al (2018) Analysis of cardiomyocyte clonal expansion during mouse heart development and injury. Nat Commun. https://doi.org/10.1038/s41467-018-02891-z
Zhang S, Kim K, Rosen A, et al. (2011) Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network Irx3 deficient mice viable. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1106911108
Ikeda U, Minamisawa M, Koyama J (2015) Isolated left ventricular non-compaction cardiomyopathy in adults. J Cardiol 65:91–97. https://doi.org/10.1016/j.jjcc.2014.10.005
Roberts WC, Karia SJ, Ko JM et al (2011) Examination of isolated ventricular noncompaction (hypertrabeculation) as a distinct entity in adults. Am J Cardiol 108:747–752. https://doi.org/10.1016/j.amjcard.2011.04.027
Costa MW, Guo G, Wolstein O et al (2014) Functional characterization of a novel mutation in NKX2-5 associated with congenital heart disease and adult-onset cardiomyopathy. Circ Cardiovasc Genet 6:1–21. https://doi.org/10.1161/CIRCGENETICS.113.000057.Functional
Ashraf H, Pradhan L, Chang EI et al (2014) A mouse model of human congenital heart disease high incidence of diverse cardiac anomalies and ventricular noncompaction produced by heterozygous Nkx2-5 homeodomain missense mutation. Circ Cardiovasc Genet 7:423–433. https://doi.org/10.1161/CIRCGENETICS.113.000281
Gittenberger-de Adriana C, Groot Hoppenbrouwers T, Miquerol L et al (2016) 14-3-3epsilon controls multiple developmental processes in the mouse heart. Dev Dyn 245:1107–1123
Choquet C, Marcadet L, Beyer S et al (2016) Segregation of central ventricular conduction system lineages in early SMA+ cardiomyocytes occurs prior to heart tube formation. J Cardiovasc Dev Dis 3:2. https://doi.org/10.3390/jcdd3010002
Bressan M, Liu G, Mikawa T (2013) Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science 340:744–748. https://doi.org/10.1126/science.1232877
Finsterer J, Stöllberger C, Towbin JA (2017) Left ventricular noncompaction cardiomyopathy: cardiac, neuromuscular, and genetic factors. Nat Rev Cardiol 14:224–237. https://doi.org/10.1038/nrcardio.2016.207
Klaassen S, Probst S, Oechslin E et al (2008) Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 117:2893–2901. https://doi.org/10.1161/CIRCULATIONAHA.107.746164
Freedom RM, Yoo SJ, Perrin D et al (2005) The morphological spectrum of ventricular noncompaction. Cardiol. Young 15:345–364
Chen H, Zhang W, Sun X et al (2013) Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 140:1946–1957. https://doi.org/10.1242/dev.089920
Luxán G, Casanova JC, Martínez-Poveda B et al (2013) Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med 19:193–201. https://doi.org/10.1038/nm.3046
Zhao C, Guo H, Li J et al (2014) Numb family proteins are essential for cardiac morphogenesis and progenitor differentiation. Development 141:281–295. https://doi.org/10.1242/dev.093690
Pashmforoush M, Lu JT, Chen H et al (2004) Nkx2-5 pathways and congenital heart disease: loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117:373–386. https://doi.org/10.1016/S0092-8674(04)00405-2
Liu Y, Chen H, Shou W (2018) Potential common pathogenic pathways for the left ventricular noncompaction cardiomyopathy (LVNC). Pediatr Cardiol 39:1099–1106. https://doi.org/10.1007/s00246-018-1882-z
Li D, Hallett MA, Zhu W et al (2011) Dishevelled-associated activator of morphogenesis 1 (Daam1) is required for heart morphogenesis. Development 138:303–315. https://doi.org/10.1242/dev.055566
Jensen B, van der Wal AC, Moorman AFM, Christoffels VM (2017) Excessive trabeculations in noncompaction do not have the embryonic identity. Int J Cardiol 227:325–330. https://doi.org/10.1016/j.ijcard.2016.11.089
Jay PY, Harris BS, Maguire CT et al (2004) Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest 113:1130–1137. https://doi.org/10.1172/JCI200419846
Meysen S, Marger L, Hewett KW et al (2007) Nkx2.5 cell-autonomous gene function is required for the postnatal formation of the peripheral ventricular conduction system. Dev Biol 303:740–753. https://doi.org/10.1016/j.ydbio.2006.12.044
Palomino Doza J, Salguero-Bodes R, de la Parte M, Arribas-Ynsaurriaga F (2018) Association Between Mutations in the NKX2.5 Homeobox, Atrial Septal Defects, Ventricular Noncompaction and Sudden Cardiac Death. Rev Española Cardiol 71:53–55. https://doi.org/10.1016/j.rec.2017.02.032
D’Amato G, Luxán G, del Monte-Nieto G et al (2015) Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol 18:7–20. https://doi.org/10.1038/ncb3280
Funding
CC is supported by the GRRC/SFC (Groupe de Réflexion sur la Recherche Cardiovasculaire/Société Française de Cardiologie), RGK is supported by the Fondation pour la Recherche Médicale (Grant #DEQ20150331717), LM by the Association Française contre les Myopathies (Grant #15619), and the Ligue contre la Cardiomyopathie.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflicts of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Animal procedures were approved by the ethics committee for animal experimentation of the French ministry (No. 01055.02).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Choquet, C., Kelly, R.G. & Miquerol, L. Defects in Trabecular Development Contribute to Left Ventricular Noncompaction. Pediatr Cardiol 40, 1331–1338 (2019). https://doi.org/10.1007/s00246-019-02161-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-019-02161-9